15845
Views & Citations14845
Likes & Shares
Alzheimer’s
disease (AD) presents amyloid plaques as one of its earliest and most
characteristic hallmarks, occurring up to 20 years before clinical diagnosis.
Their precise role in the onset of AD is still being investigated but they
appear to be an effective biomarker of its pre clinical stages. These plaques
seem to be part of the neurodegeneration process in AD that leads to the
progressive deterioration of glia and neurons in specific brain regions.
Working upon the hypothesis that the biological response to neurodegeneration
induces the development and massive generation of pathological hallmarks in the
brain, AD-related hallmark immune-expression was analyzed in transverse brain sections
of two transgenic mouse models. A newly-generated triple-transgenic mouse
(APP/BIN1/COPS5) was compared to double-transgenic mice (APP/PS1) using
immunohistochemistry detection methods. A comparison of disease-specific
hallmark changes and neuropathological biomarkers throughout disease evolution
revealed a different hallmark pattern in the two models, providing novel
insight into the development of AD pathology. This study presents for the first
time an age-related comparative pattern of the neuropathological framework of
AD in transgenic mouse models, key to understanding the genetic-specific
targets for immunotherapy and neuronal protection.
INTRODUCTION
Epigenomic
regulation is a universal phenomenon of gene expression control during
development, maturation and aging in physiological conditions. When this
mechanism of controls altered by endogenous and/or exogenous factors, probably
acting as an interface between the genome and the environment (nature vs
nurture) [1,2], then epigenomic changes become pathogenic due to the abnormal
expression of genes under epigenetic control. Harris et al. [3] define
these metastable epialleles
as mammalian genomic loci where epigenetic patterning occurs before
gastrulation in a stochastic fashion, leading to systematic interindividual
variation within one species. This gene expression abnormally leads to a
potential reversible pathological phenotype which, in some cases, can be
transferred to future generations, assuming that epigenetics refers to
phenotypic changes with no apparent alterations in structural DNA. Preconceptional
parental exposure to environmental stimuli may determine the offspring’s
phenotype via meiotically and mitotically heritable epigenetic mechanisms [1],
and exposure to diverse external elements (nutrition, pollutants, drugs,
toxins) may condition several categories of human diseases. Classical
epigenetic mechanisms, including DNA methylation, histone modifications, and
regulation by microRNAs (miRNAs), are among the major regulatory elements that
control metabolic pathways at the molecular level. DNA
methylation/demethylation and chromatin remodeling/histone modifications
regulate gene expression transcriptionally, and miRNAs suppress gene expression
post-transcriptionally [4]. Mutations in the genes encoding elements of the
epigenetic machinery can lead to an epigenetic Mendelian disorder [5].
Epigenetic marks contribute to natural human variation [6] and
configure the emerging field of neuroepigenetics [2]. Not only nuclear DNA, but
also mitochondrial DNA may be subjected to epigenetic modifications related to
disease development, environmental exposure, drug treatment and aging [7]. Some
epigenetic modifications are conceptually reversible and can potentially be
targeted by pharmacological and dietary interventions [8-10].
Age-related neuropsychiatric disorders (from neurodevelopment to aging)
are complex diseases in which genomic defects, together with environmental
factors and epigenetic alterations, may be involved [11]. Most of these
disorders exhibit proteoepigenomic changes resulting from primary genomic
traits and/or secondary epigenetic events which induce pathogenic (structural,
functional, conformational) changes in key proteins [12]. Consequently,
neuroepigenetic perturbations in genes involved in brain development,
maturation and aging, may alter gene expression and protein synthesis (and
conformational protein configuration) leading to neurodevelopmental,
neuropsychiatric, and neurodegenerative disorders [13] (Table 1). Epigenetic changes in genes involved in pharmacogenomics
(pathogenic, mechanistic, metabolic, transporter, and pleiotropic genes) can
also influence drug efficacy and safety and drug resistance in brain disorders
and cancer [14].
EPIGENETIC
MECHANISMS
DNA methylation
The human genome may contain approximately ∼29
million CpG dinucleotides, and the number of potential methylation patterns per
haploid genome might be around 108,700,000, this contributing to
increase the information content of the genome and endowing mammalian genomes
with the ability to subjugate specific sequences to irreversible
transcriptional silencing [15]. Methylation varies spatially across the genome,
with a majority of the methylated sites mapping to intragenic regions [16]. DNA
methylation is a process by which methyl groups are incorporated into cytosine
molecules by DNA methyltransferases (DNMTs), forming 5-methylcytosine and
contributing to the suppression of transcription. Approximately 70% of CpG
dinucleotides within the human genome are methylated. CpG islands in promoter
regions of genes are defined as 200 bp regions of DNA where the GC content is
greater than 60%. DNA methylation inhibits transcription by interfering with
the binding of transcription factors to recognition sites on promoters or by
recruiting and binding transcriptional repressors, methyl-CpG-binding proteins
(MBDs), and altering chromatin structure into an active state.
5-Methylcytosines (5mC) can also be oxidized to form 5-hydroxymethylcytosine
(5hmC) to reduce the interaction of DNA with DNA-binding proteins [17]. CpG
methylation may also cause a dual effect on transcription, repressing
transcription when CpG methylation occurs at the promotor level or promoting
transcription when CpG methylation affects the gene sequence [18]. A family of
DNMTs catalyzes the transfer of methyl groups from S-adenosyl-methionine (SAM)
to cytosine in CpGs. In mammals there are 2 de
novo DNMTs (DNMT3A, DNMT3B) and a maintenance DNMT (DNMT1) that is
expressed in neurons. DNMT2 methylates aspartic acid tRNA, and does not
methylate DNA [19,20]. DNA demethylation can be produced by at least 3 enzyme
families: (i) the ten-eleven translocation (TET) family, mediating the conversion
of 5mC into 5hmC; (ii) the AID/APOBEC family, acting as mediators of 5mC or
5hmC deamination; and (iii) the BER (base excision repair) glycosylase family
involved in DNA repair [17]. Lysine-Specific Demethylase 1 (LSD1) (also known
as KDM1A and AOF2) is a histone modifier involved in transcriptional
repression, forming a stable core complex with the corepressor of REST (CoREST)
and histone deacetylases (HDAC1/2) [9,10].
Non-CG methylation (mCH) is abundant and nonrandomly distributed in the
genomes of pluripotent cells and brain cells, and is present at lower levels in
many other human cells and tissues. mCH in pluripotent cells is distinct from
that in brain cells in terms of sequence specificity and association with
transcription, indicating the existence of different mCH pathways. In brain
cells, mCH accumulates during the establishment of neural circuits and is
associated with Rett syndrome [21].
Histone
modifications
Histone modifications (HMs) (histone acetylation, methylation,
phosphorylation, sumoylation, ubiquitylation, glycosylation, ADP ribosylation,
biotinylation) are essential epigenetic features, with fundamental roles in
biological processes such as transcription, DNA repair and DNA replication.
[17,22]. Histone acetylation is achieved by the action of histone
acetyltransferase (HAT), which adds an acetyl group to a lysine residue,
resulting in chromatin/transcriptional activation; histone deacetylation is
produced by histone deacetylases (HDACs) which remove the acetyl groups, and is
related to chromatin inactivation and transcriptional repression [22,23].
Chromatin
remodeling: Stable
heterochromatin is necessary to silence transposable elements (TEs) and
maintain genome integrity. Chromatin regulators (CRs) mediate HMs to adjust
chromatin structures and functions. ATP-dependent chromatin remodeling
complexes (ACRCs) use ATP hydrolysis to move, destabilize, eject or restructure
nucleosomes, allowing the accessibility of transcription factors to DNA. There
are at least 4 families of ACRCs: (i) the SWI/SNF (switching defective/sucrose
nonfermenting) family; (ii) the ISWI (imitation SWI) family; (iii) the CHD
(chromodomain, helicase, DNA binding) family; and (iv) the INO (inositol
requiring 80 family) [24]. Their transcriptional effects (activation or
repression) depend upon the recruitment of coactivators or corepressors [17].
Post-translational
histone modifications:Post-translational histone changes include acetylation, ubiquitylation,
or sumoylation at K (lysine) residues, methylation at K, R (arginine) or H
(histidine) residues, and phosphorylation at S (serine), T (threonine) or Y
(tyrosine) residues, which affect transcription, DNA replication, and DNA
repair [17].
Histone acetylation is catalyzed by 5 families of histone lysine
acetyltransferases (KATs) (KAT2A/GCN5, KAT2B/PCAF, KAT6-8, CREBBP/CBP, EP300)
[25]. Histone acetylation is associated with transcriptional activation and
open chromatin conformation. In contrast, histone deacetylation is involved in
transcriptional repression and closed chromatin structure. 18 HDACs, present in
mammals, are organized into 4 classes (class I, II, III, IV): (i) Class I HDACs
(HDAC1, 2, 3, and 8), nuclear proteins; HDAC1 and HDAC2 are often found in
transcriptional corepressor complexes (SIN3A,
NuRD, CoREST), and HDAC3 is found in other complexes (SMRT/N-CoR); (ii)
class II HDACs are subdivided into class IIa (HDAC4, 5,7 and 9), and IIb (HDAC6
and 10), which are located in the nucleus-cytoplasm interface and in the
cytoplasm, respectively; (iii) class III HDCAs belong to the sirtuin (SIRT)
family, with nuclear (SIRT1, 2, 6, 7), mitocondrial (SIRT3, 4, 5), or
cytoplasmic (SIRT1, 2) localization; and (iv) class IV HDAC (HDAC11), a nuclear
protein. HDACs regulate gene expression by inducing conformational changes in
chromatin [9,10,17,22,26,27]. H3 (K9, K14, K18, K56), H4 (K5, K8, K12, K16), and
H2B (K6, K7, K16, K17) acetylation, H3 (K4me2, K4me3, K36me3, K79me2)
methylation, and H3 (S10) phosphorylation activate transcription, and H3
(K9me3, K27me3) and H4 (K20me3) methylation represses transcription [28].
Non-coding RNAs
Over 95% of the eukaryotic genome is transcribed into non-coding RNAs
(ncRNAs) and less than 5% is translated [29,30]. Long non-coding (lnc) RNAs are
non-protein-coding RNAs, distinct from housekeeping RNAs (tRNAs, rRNAs, and
snRNAs) and independent from small RNAs with specific molecular processing
machinery (micro- or piwi-RNAs) [31]. Long RNA (lncRNA)-mediated epigenetic
regulation depends mainly on lcnRNA interactions with proteins or genomic DNA
via RNA secondary structures. ncRNAs are classified by size into 2 categories:
(i) small RNAs (<200 nucleotides): (a) structural RNAs: ribosomal (rRNA),
transfer (tRNA), small nuclear RNAs (snRNA); (b) regulatory RNAs: microRNAs
(miRNA), small interfering RNAs (siRNA), small nuclear RNAs (snRNA),
piwi-interacting RNAs (piRNA), splice junction-associated RNAs; and (ii) long
RNAs (lncRNAs) (>200 nucleotides), present in >8000 loci in the human
genome: large intergenic non-coding RNAs (lincRNA), natural antisense
transcripts (NATs), non-coding RNA expansion repeats, promoter-associated RNAs
(PARs), enhancer RNAs (eRNAs), small activating RNAs (saRNAs, RNAa) [17,32,33].
Small non-coding RNAs (ncRNAs) -miRNAs, siRNAs, piRNAs- show mature forms of
20-30 nucleotides (nt) that associate with members of the Argonaute (AGO)
superfamily of proteins, the central effectors of RNA interference (RNAi)
pathways. miRNAs and siRNAs are post-transcriptional gene silencers, guiding
AGO complexes to complementary mRNAs in the cytoplasm, inducing transcript
degradation and blocking translation [32]. miRNAs repress translation with RISC
(RNA-induced silencing complex) and induce mRNA degradation by binding to the
3’ untranslated region (3’UTR). Other miRNAs may enhance mRNA translation and
induce gene expression by binding to the promoter of the target gene. ncRNAs
are essential in the regulation of epigenetic mechanisms (silencing of
transposable elements, gene expression control, X-chromosome inactivation, DNA
imprinting, DNA methylation, histone modifications). piRNAs are essential for
fertility, associating with the PIWI clade of Argonautes to silence transposons
in the germline [32]. RNA activation (RNAa) is currently accompanied by changes
in histone modifications around the target promoter, and DNA methylation does
not appear to be affected by RNAa [32], although RNA-directed DNA methylation
(RdDM) and RNA-induced transcriptional silencing (RITS) phenomena have been
reported [34]. Endogenous small RNA-mediated epigenetic gene regulation
involves miRNA-induced RNAa and miRNA-induced transcriptional gene silencing
[34].
NATs are lncRNAs arising from the opposite strand of protein-coding or
non-protein-coding genes that regulate mRNA expression at the level of
transcription via competition for regulatory factors, or through physically
hindering the progress of transcription. NATs edit or activate cellular
siRNA-related pathways that lead to degradation of homologous transcripts,
ultimately eliciting gene silencing and can regulate RNA processing including
translation, polyadenylation, splicing, transport or degradation. NATs can also
bind to epigenetic enzymes and act as a scaffold to form active or repressive
chromatin modifying complexes [35]. NATs have been associated with
neurodegenerative, neurodevelopmental and psychiatric disorders (schizophrenia,
bipolar disorder, autism, and fragile X mental retardation gene (FMR1) [36].
The lncRNA Xist initiates X chromosome inactivation (Xi) in female
somatic cells, silencing a number of genes on the inactive X chromosome,
necessary for a normal brain development [37]. LncRNAs also regulate gene
expression through genomic imprinting [38]. LncRNAs can also regulate gene
expression through interaction with paraspeckles, membraneless subnuclear
bodies that participate in nuclear organization, regulating gene expression post-transcriptionally.
The formation and maintenance of paraspeckles requires NEAT1, a lncRNA that localizes exclusively to paraspeckles. NEAT1
is upregulated in Huntington’s disease [39] and in amyotrophic lateral
sclerosis (ALS) [40].
Brain Development
Dynamic epigenetic changes are fundamental for normal brain
development. All components of the epigenetic machinery participate in the
normal process of brain development. Mendelian mutations in different
epigenetic factors cause irreversible neurodevelopmental and imprinting
disorders. DNA methylation is a mechanism of epigenetic control in mammals at
different stages of the lifespan. During embryonic development, DNA methylation
restricts differentiation and prevents regression into an undifferentiated state,
compensates sex chromosome dosage, represses retrotransposons that threaten
genome integrity, maintains genome stability, and coordinates the expression of
imprinted genes [41]. Vertebrate genomes undergo epigenetic reprogramming
during development and disease. Stable transmission of DNA methylation,
transcriptomes and phenotypes from parent to clonal offspring are demonstrated
in various asexual species, and clonal genotypes from natural populations show
habitat-specific DNA methylation [42]. Prenatal exposure to deleterious factors
may induce aberrations in DNA methylation and other epigenetic mechanisms,
leading to the abnormal expression of genes with negative effects on
neurodevelopment, experience-dependent plasticity, brain sex differentiation and
brain maturation later in life [13,43]. There is evidence that adverse effects
of early-life stress are pervasive, with well-established mental and physical
health consequences for exposed individuals. The impact of early adverse
experiences is also highly persistent, with documented increases in risk for
mental illness across the lifespan. Stress phenotypes may persist even beyond
the lifespan of the individual, with consequences for their offspring and
grand-offspring. Phenotypic characteristics may be transmitted to future
generations via either the matriline or the patriline, a phenomenon that has
been demonstrated in both human and animal studies [44].
Regulation of specialized genes might also be a form of epigenetic
predestination with effects on brain evolution, development and maturation.
Sushi-ichi-related retrotransposon homolog 11/Zinc finger CCHC
domain-containing 16 (Sirh11/Zcchc16)
encodes a CCHC type of zinc-finger protein that exhibits high homology to an
LTR retrotransposon Gag protein. Sirh11/Zcchc16
is involved in cognitive function and gene targeting of mouse Sirh11/Zcchc16 causes abnormal behaviors
(cognition deficits: attention, impulsivity and working memory). This gene is
highly conserved in the eutherians (euarchontoglires, laurasiatheria and
afrotheria) and is heavily mutated in xenarthran species such as the sloth and
armadillo, suggesting that it has contributed to brain evolution in the three
major eutherian lineages, including humans and mice. According to data reported
by Irie et al. [45], Sirh11/Zcchc16
is the first SIRH gene to be involved in brain function, instead of the
placenta, as seen in the case of Peg10,
Peg11/Rtl1 and Sirh7/Ldoc1.
The regulation of methylation/demethylation pathways in the central
nervous system (CNS) is highly controlled, depending on brain region and age
[46]. 5-Hydroxymethylcytosine (5hmC) is an oxidative product of
5-methylcytosine (5mC), catalyzed by the TET family of enzymes. These enzymes
are thought to play a role in mammalian development and differentiation. TET
enzymes are mutated in several types of cancer, affecting their activity and
likely altering genomic 5hmC and 5mC patterns. Oxidation of 5mC appears to be a
step in several active DNA demethylation pathways, which may be important for normal
processes, as well as global hypomethylation during cancer development and
progression [47]. 5hmC is also involved in Rett syndrome [48].
Epigenetic modifications of histone proteins and DNA seem to be a
leading molecular mechanism to modulate the transcriptional changes underlying
the fine-tuning of synaptic connections and circuitry rewiring during
activity-dependent plasticity [13]. Many lncRNAs are expressed in the CNS where
they participate in essential processes for normal brain development [33,49].
Age-related
Epigenetics
Altered DNA methylation patterns may account for phenotypic changes
associated with human aging. Brain region-specific expression of genes can be
epigenetically regulated by DNA methylation [50] and brain aging might be
influenced by epigenetic changes in the neuronal microenvironment [51,52].
DNA Methylation
Age- and tissue-dependent DNA hypo- and hypermethylation has been
reported [17]. It appears that global loss of DNA methylation predominates in
aged cells. DNMT1, which maintains DNA methylation of CpGs, decreases with age
[53]. In contrast, some loci have been found hypermethylated with age (e.g.
estrogen receptor, interferon γ, insulin-like growth factor II, promoters of tumor-suppressor genes
such as lysyl oxidase (LOX), p16INK4a, runt-related transcription
factor 3 (RUNX3), and TPA-inducible
gene 1 (TIG1)) [17]. Xu and Taylor
[54] analyzed 1,006 blood DNA samples of women aged 35 to 76 from the Sister
Study, and found that 7,694 (28%) of the 27,578 CpGs assayed were associated
with age, confirming the existence of at least 749 "high-confidence"
age-related CpG (arCpGs) sites in normal blood. These age-related changes are
largely concordant in a broad variety of normal tissues, and a significantly
higher (71-91%) than expected proportion of increasingly-methylated arCpGs
(IM-arCpGs) were over-methylated in a wide variety of tumor types. IM-arCpGs
sites occurred almost exclusively at CpG islands and were disproportionately
marked with the repressive H3K27me3 histone modification. These findings
suggest that as cells acquire methylation at age-related sites they have a
lower threshold for malignant transformation that may explain in part the
increase in cancer incidence with age.
McClay et al. [55] performed a methylome-wide association study of
aging in whole blood DNA from 718 individuals, aged 25-92 years. They sequenced
the methyl-CpG-enriched genomic DNA fraction, averaging 67.3 million reads per
subject, to obtain methylation measurements for the ∼27
million autosomal CpGs in the human genome, and adaptively combined methylation
measures for neighboring, highly-correlated CpGs into 4,344,016 CpG blocks for
association testing. Eleven age-associated differentially methylated regions
(DMRs) passed Bonferroni correction. 42 of 70 selected DMRs showed
hypomethylation and 28 showed hypermethylation with age. Hypermethylated DMRs
were more likely to overlap with CpG islands and shores. Hypomethylated DMRs
were more likely to be in regions associated with polycomb/regulatory proteins
(EZH2) or histone modifications H3K27ac, H3K4m1, H3K4m2, H3K4m3 and H3K9ac. Among
genes implicated by the top DMRs were protocadherins, homeobox genes,
mitogen-activated protein kinases (MAPKs), ryanodine receptors, and genes with
potential relevance for age-related disease.
The absolute levels of 5-hydroxymethylcytosine (hmC), 5-formylcytosine
(fC) and 5-methylcytosine (mC) vary in human brain tissues at various ages. For
hmC, an initial steady increase is observed, which levels off with age to a
final steady-state value of 1.2%. This level is nearly twice as high as in
mouse cerebral cortex. fC declines rapidly with age during early developmental
stages. While hmC is a stable epigenetic mark, fC is more likely an
intermediate of active DNA demethylation during early brain development. The
trends in global cytosine modification dynamics during the lifespan are
conserved between humans and mice and show similar patterns in different organs
[56].
Histone
modifications
Histone modifications are also observed with aging. Histone acetylation
decreases and phosphorylation increases with age [57]. H4K20me and H3K36me3
decrease in the brain of old senescence-accelerated prone mice (SAMP8) and
H3K27m3, H3K79me, and H3K79me2 increase in these aged mice brains [58]. The
silent information regulator 2 (Sir2) in yeast and its mammalian orthologs, sirtuin
1-7 (SIRT1-7), are histone-modifying enzymes which tend to be dowregulated in
aging, especially SIRT1. Activation of sirtuins may extend lifespan, modulating
calorie restriction mechanisms [59] and promoting a healthy aging, which delays
the onset of neurodegenerative processes [60]. In the epidermis, aging is
associated with a limited destabilization of the epigenome at gene regulatory
elements [61]. Wound treatment with Sirtuin activators and class I HDAC
inhibitors induce keratinocyte proliferation and enhances healing via a nitric
oxide (NO)-dependent mechanism. Acetylation of α-tubulin and histone H3 Lysine 9
may activate cell function and gene expression to foster tissue repair. The
direct activation of P300/CBP-associated
factor (PCAF) by the histone acetylase activator pentadecylidenemalonate 1b
(SPV-106) induces lysine acetylation in the wound area. An impairment of PCAF
and/or other GCN5 family acetylases may delay skin repair in physiopathological
conditions [62].
Non-coding DNAs
There is a correlation between changes in miRNA expression and aging.
miRNA lin-4 regulates lifespan in C.
elegans; several miRNAs (miRNAs-34, -669c, -709, -93, -214) were found to
be upregulated with age, while others (miRNAs-103, -107,-128, -130a, -155, -24,
-221, -496, -1538, -17, -19b, -20ª, -106a) appeared downregulated in peripheral
tissues [63,64]. 70 miRNAs were found to be upregulated in the aging brain; 27
of these miRNAs may target genes of mitocondrial complexes III, IV, and F0F1-ATPase
involved in oxidative phosphotylation and reduced expression in aging [65].
Neurodevelopmental
Disorders
Epigenetic mechanisms are determinant in brain development and
maturation, puberty-related changes [66], mental disorders [67-70], addictive
behaviors [71,72], and neurodegeneration [14,17,73,74].There is a number of
neurodevelopmental disorders in which epigenetic dysregulation plays an
important role (autism spectrum disorders, Rett syndrome, fragile X syndrome,
Prader-Willi syndrome, Angelman syndrome, and Kabuki syndrome) [75,76] (Table 1).
Methylation of histone H3 lysine 4 (H3K4me) is a regulated
post-translational modification, which is broadly associated with enhancers and
promoters of actively transcribed genomic loci. Four H3K4me methyltransferases
(KMT2A, KMT2C, KMT2D, KMT2F), four demethylases (KDM1A, KDM5A, KDM5B, KDM5C),
and two reader proteins (PHF21A, PHF8) are mutated in neurodevelopmental
disorders [77].
Rett syndrome is an X-linked neurodevelopmental disease caused by MECP2 mutations. The MeCP2 protein acts
as a transcription repressor by binding to methylated CpG dinucleotides, and
also as a transcription activator. MeCP2 is expressed in neurons and in glial
cells. Reintroduction of MeCP2 into behaviorally affected Mecp2-null mice after birth rescues neurological symptoms,
indicating that epigenetic failures in Rett syndrome are reversible [78].
Mutations in JMJD1C (jumonji domain containing 1C) contribute to the
development of Rett syndrome and intellectual disability (ID). Mutant JMJD1C in
Rett syndrome has abnormal subcellular localization, diminished activity to
demethylate the DNA damage-response protein MDC1, and reduced binding to MECP2
[79].
A body of novel arguments postulates the involvement of epigenetic
mechanisms in the pathogenesis of autism [80-82]. Autism spectrum disorders
(ASD) are a heterogeneous group of neurodevelopmental disorders which are
comorbid with attention deficit hyperactivity disorder (ADHD), epilepsy, Rett
syndrome, and Fragile X syndrome [83,84]. There are several epigenome-wide
association studies of ASD suggesting a potential role for epigenetics in ASD
pathogenesis [82].
Mbadiwe and Millis [80] reviewed mechanisms for altering DNA-histone
interactions of cell chromatin to upregulate or downregulate gene expression
that could serve as epigenetic targets for therapeutic interventions. The
proposed rationale includes the following sequence: (i) DNA methyltransferases
(DNMTs) phosphorylate histone H3 at T6; (ii) the DNMT lysine-specific demethylase-1
prevents demethylation of H3 at K4; (iii) during androgen-receptor
(AR)-dependent gene activation, this sequence may produce AR-dependent gene
overactivation which may partly explain the male predominance of autism; (iv)
AR-dependent gene overactivation in conjunction with a DNMT mechanism for
methylating oxytocin receptors could produce high arousal inputs to the
amygdala resulting in aberrant socialization, a prime characteristic of autism;
(v) dysregulation of histone methyltransferases and histone deacetylases
(HDACs) associated with low activity of methyl CpG binding protein-2 at
cytosine-guanine sites in genes may reduce the capacity for condensing
chromatin and silencing genes in frontal cortex, a site characterized by
decreased cortical interconnectivity in autistic subjects; and (vi) HDAC1
inhibition can overactivate mRNA transcription, a putative mechanism for the
increased number of cerebral cortical columns and local frontal cortex
hyperactivity in autistic individuals [80,85].
Sullivan et al. [86] identified the bromodomain and extraterminal
domain-containing proteins (BETs) as epigenetic regulators of genes involved in
ASD-like behaviors in mice. The pharmacological suppression of BET proteins in
the brain of young mice, by the novel, brain-permeable inhibitor I-BET858 leads
to selective suppression of neuronal gene expression followed by the
development of an autism-like syndrome. Many of the I-BET858-affected genes
have been linked to ASD in humans, suggesting the key role of the BET-controlled
gene network in the disorder.
A genome-wide differential expression of long noncoding RNAs (lncRNAs)
was identified in blood specimens of ASD. A total of 3929 lncRNAs were found to
be differentially expressed in ASD peripheral leukocytes, including 2,407 that
were upregulated and 1,522 that were downregulated. Simultaneously, 2,591
messenger RNAs (mRNAs), including 1,789 upregulated and 821 downregulated, were
also identified in ASD leukocytes. Functional pathway analysis of these lncRNAs
revealed neurological pathways of the synaptic vesicle cycling, long-term
depression and long-term potentiation to be primarily involved. Thirteen
synaptic lncRNAs, including 9 upregulated and 4 downregulated, and 19 synaptic
mRNAs, including 12 upregulated and 7 downregulated, were identified as being
differentially expressed in ASD. Discovery of the lncRNAs SHANK2-AS and BDNF-AS,
the natural antisense of genes SHANK2 and BDNF, respectively, indicates that in
addition to gene mutations, deregulation lncRNAs on ASD-causing gene loci
presents a new approach for exploring possible epigenetic mechanisms underlying
ASD [87].
Fragile X syndrome (FXS) is the most common monogenic form of
developmental cognitive impairment. FXS represents a prototype of the so-called
repeat expansion disorders due to "dynamic" mutations of a CGG repeat
in the 5'UTR of the FMR1 gene. This
genetic anomaly is accompanied by epigenetic modifications (mainly DNA
methylation and histone deacetylation), resulting in the inactivation of the FMR1 gene. The presence of an intact FMR1 coding sequence allowed
pharmacological reactivation of gene transcription, particularly through the
use of the DNA-demethylating agent 5'-aza-2'-deoxycytydine and/or inhibitors of
histone deacetylases. These treatments suggested that DNA methylation is
dominant over histone acetylation in silencing the FMR1 gene. The importance of DNA methylation in repressing FMR1 transcription is confirmed by the
existence of rare unaffected males carrying unmethylated full mutations [88].
The 22q11.2 deletion syndrome (22qDS), with a hemizygous deletion of
1.5-3 Mb on 22q11.2, is the most common microdeletion disorder (prevalence:
1/4000) and the second risk factor for schizophrenia. At least 9 (COMT, UFD1L, DGCR8, MRPL40, PRODH, SLC25A1, TXNRD2,
T10, and ZDHHC8) of 30 genes involved in 22qDS have the potential of
disrupting mitochondrial metabolism. Deficits in bioenergetics during early
postnatal brain development may set the basis for a disrupted neuronal
metabolism or synaptic signaling. Altered metabolism in 22qDS reflects a
critical role for the haploinsufficiency of the mitochondrial citrate
transporter SLC25A1, further enhanced
by HIF-1α, MYC, and metabolite controls [89].
Imprinting disorders
Epigenetic regulation of imprinted genes during embryonic development
is influenced by the prenatal environment [90]. Genomic imprinting refers to an
epigenetic mark that distinguishes parental alleles and results in a
monoallelic, parental-specific expression pattern in mammals. The alleles of imprinted
genes are marked epigenetically at discrete elements termed ‘imprinting control
regions’ (ICRs) with their parental origin in gametes through the use of DNA
methylation. Imprinted gene expression is subsequently maintained using
noncoding RNAs, histone modifications, insulators, and higher-order chromatin
structure. Avoidance is manifest when imprinted genes evade the genome-wide
reprogramming that occurs after fertilization and remain marked with their
parental origin [91].
DNA methylation is a hallmark of genomic imprinting and differentially
methylated regions (DMRs) are found near and in imprinted genes. Imprinted
genes are expressed only from the maternal or paternal allele and their normal
balance can be disrupted by uniparental disomy (UPD), i.e., the inheritance of
both chromosomes of a chromosome pair exclusively from only either the mother
or the father. A growing number of congenital disorders have been linked to
genomic imprinting. Each of these is caused by perturbed gene expression at one
principal imprinted domain. Some imprinting disorders, including the
Prader-Willi and Angelman syndromes, are caused almost exclusively by genetic
mutations, although hypermethylation at the ICRs may also contribute to the
maternal or paternal allele silencing. In other cases, including the
Beckwith-Wiedemann and Silver-Russell growth syndromes, and transient neonatal
diabetes mellitus, imprinted expression is perturbed mostly by epigenetic
alterations at ICRs and at other specific regulatory sequences. In a minority
of these patients, DNA methylation is altered at multiple imprinted loci,
suggesting that common trans-acting factors are affected [92].
Maternal UPD for chromosome 7 (matUPD7) results in Silver-Russell
syndrome (SRS) with typical features and growth retardation, but no gene has
been conclusively implicated in SRS. Genome-scale analysis of eight matUPD7
patients, a segmental matUPD7q31-qter,
a rare patUPD7 case, and ten controls on the Infinium HumanMethylation450K
BeadChip with 30,017 CpG methylation probes for chromosome 7 showed highly
significant clustering of DMRs only on chromosome 7, including the known
imprinted loci GRB10, SGCE/PEG10, and PEG/MEST. Ten novel DMRs on chromosome
7, two DMRs for the predicted imprinted genes HOXA4 and GLI3, and one
for the disputed imprinted gene PON1,
and differential expression for three genes with novel DMRs, HOXA4, GLI3, and SVOP, were also demonstrated. Allele-specific expression analysis
confirmed maternal only expression of SVOPL and imprinting of HOXA4 was supported by monoallelic
expression. These results reported by Hannula-Jouppi et al. [93] represent the
first comprehensive map of parent-of-origin- specific DMRs on human chromosome
7, suggesting many new imprinted sites.
Kagami-Ogata syndrome (KOS14) is another imprinting disorder caused by
an epimutation (hypermethylation) of two DMRs functioning as imprinting control
regions, namely, IG-DMR and MEG3-DMR [94].
Psychiatric
Disorders
Gene-specific and genome-wide studies of postmortem brain and blood
cells indicate that aberrant DNA methylation, histone modifications and
dysregulation of miRNAs are linked to the pathogenesis of mental diseases [95]
(Table 1). Human exome sequencing
and genome-wide association studies have linked several neurobiological
disorders to genes whose products actively regulate DNA methylation and histone
acetylation. Nucleosome remodeling has been implicated in human developmental
and intellectual disability disorders. Nucleosome remodeling is driven
primarily through nucleosome remodeling complexes with specialized
ATP-dependent enzymes. These enzymes directly interact with DNA or chromatin
structure, as well as histone subunits, to restructure the shape and
organization of nucleosome positioning and ultimately regulate gene expression.
Mutations in genes of the the neuron-specific Brg1/hBrm Associated Factor (nBAF)
complex subunit have been linked to Coffin-Siris syndrome (CSS),
Nicolaides-Baraitser syndrome (NBS), schizophrenia (SCZ), and Autism Spectrum
Disorder (ASD) [96].
Quality of maternal care experienced during infancy is a key factor
that can confer vulnerability or resilience to psychiatric disorders later in
life. Experiences within an adverse caregiving environment produce aberrant DNA
methylation patterns at various gene loci in the medial prefrontal cortex of
developing and adult experimental animals [97]. These particular conditions may
alter the network of genes involved in mental activity whose region- and
neurochemical pathway-specific dysregulation might lead to the future onset of
mental disorders.
Schizophrenia
Schizophrenia (SCZ) is a neurodevelopmental heritable disorder
(80–85%), in which hundreds of dysfunctional genomic regions are involved, with
a high rate of monozygotic concordance [69,73,98,99]. Disruption of epigenetic
processes may play an important role in the development of SCZ [100,101];
however, SCZ DNA methylation biomarkers in blood did not yield any conclusive
result [102]. The application of padlock probe-based ultra-deep bisulfite sequencing
for fine mapping of modified cytosines of the HLA complex group 9 gene in the
postmortem brains of individuals affected with SCZ or bipolar disorder and
unaffected controls detected significant differences between patients and
controls in both CpG and CpH modifications, with epigenetic age effects [103].
Methylation of DNA repetitive sequences (LINE-1
and BAGE) in peripheral blood
leukocytes from first-episode schizophrenia (FES) patients vs healthy controls
(HCs) indicate that FES+
patients have significantly lower LINE-1
methylation in comparison with FES- patients or HC-subjects.
Emotional abuse and total trauma score predicted lower LINE-1 methylation in FES patients, while general trauma score was
associated with lower BAGE methylation in HCs [104]. In genome-wide DNA
methylation analysis in post-mortem human brain tissue, 4,641 probes
corresponding to 2,929 unique genes were found to be differentially methylated.
Of those genes, 1,291 were located in a CpG island and 817 were in a promoter
region. These include NOS1, AKT1, DTNBP1,
DNMT1, PPP3CC and SOX10. More
than 100 of these genes overlap with a previous DNA methylation study of
peripheral blood from SCZ patients in which 27,000 CpG sites were analyzed
[105].
Molecular dysregulation in SCZ affects disruption of the dopamine,
N-methyl-D-aspartate (NMDA), and GABA signaling pathways under control of the
epigenetic machinery [106]. The integration of methylome-wide association study
results with GWAS findings replicated the top three methylation findings near
genes SDCCAG8, CREB1 and ATXN7 in an
independent sample using targeted pyrosequencing [107]. Hypomethylation at the LRRTM1 promoter, particularly of the
paternally inherited allele, may be an additional risk factor for the
development of SCZ in a set of siblings affected with familial SCZ [108].
Histone deacetylases (HDACs) are key enzymes of histone acetylation, and
abnormalities in histone modifications and in the level of HDAC proteins have
been reported in SCZ. The most significant maker associated with SCZ is rs14251
(HDAC3); however, rs17265596 (HDAC9), rs7290710 (HDAC10) and rs7634112 (HDAC11)
might also be involved to a lesser extent [109].
Men have a higher incidence of SCZ than women, with increases in
negative and cognitive symptoms, and an overall poorer disease course. SCZ is
conceptualized as a disorder of aberrant gene transcription and regulation.
Peripheral histone methyltransferase (HMT) mRNA levels have been shown to be
significantly increased in patients with SCZ and correlate with symptomology.
Men with SCZ express the highest levels of G9α,
SETDB1 mRNA and H3K9me2 protein levels. Higher levels of symptom
presentation and an overall poorer quality of life correlate with higher HMT mRNA and H3K9me2 protein levels in a
sex-dependent pattern. These data support the hypothesis of a sex-dependent
restrictive epigenome contributing towards the etiology of SCZ [110].
Downregulation of glutamic acid decarboxylase67 (GAD1), reelin (RELN), and
BDNF expression in SCZ and bipolar
disorder is associated with overexpression of DNA methyltransferase1 (DNMT1)
and ten-eleven translocase methylcytosine dioxygenase1 (TET1). Altered promoter
methylation may be one mechanism underlying the down-regulation of GABAergic
and glutamatergic gene expression. Both DNMT1 and TET1 directly bind to
unmethylated CpG-rich promoters through their respective Zinc Finger (ZF-CXXC) domains. The binding of DNMT1
to GABAergic (GAD1, RELN) and
glutamatergic (BDNF-IX) promoters is increased in SCZ and bipolar disorder and
this increase does not necessarily correlate with enrichment in promoter
methylation. Increased binding of DNMT1 positively correlates with increased
expression of DNMT1 and with increased binding of MBD2. In contrast, the
binding of TET1 to RELN, GAD1and BDNF-IX promoters failed to change [111]. Expression of GAD1 GABA synthesis enzyme is highly
regulated by neuronal activity and reaches mature levels in the prefrontal
cortex not before adolescence. Patients with SCZ show deficits in GAD1 RNA and protein levels in multiple
areas of adult cerebral cortex, possibly reflecting molecular or cellular
defects in subtypes of GABAergic interneurons essential for network
synchronization and cognition. Deficits in cortical GAD1 RNA are associated with changes in the epigenetic architecture
of the promoter, affecting DNA methylation patterns and nucleosomal histone
modifications. These localized chromatin defects at the 5' end of GAD1 are superimposed by disordered
locus-specific chromosomal conformations, including weakening of long-range
promoter-enhancer loopings and physical disconnection of GAD1 core promoter sequences from cis-regulatory elements
positioned 50 kilobases further upstream [112]. Gadd45b siRNA transfection in neurons abolishes the NMDA-induced
increase in Bdnf IXa mRNA and
reductions in 5MC and 5HMC at the Bdnf
IXa promoter in post-mytotic neurons [113]. Prenatal methylazoxymethanol
(MAM) administration impairs histone acetylation in the prefrontal cortex,
which might be involved in the development of some of the neurobehavioral
deficits associated with SCZ; and blockade of HDAC2 with valproic acid might
prevent the disruption of sensorimotor gating in adulthood [114].
Klinefelter syndrome (KS) is the most common sex-chromosome aneuploidy
in humans. Most affected individuals carry one extra X-chromosome (47, XXY
karyotype) and the condition presents with a heterogeneous mix of reproductive,
physical and psychiatric phenotypes. Genomic, methylomic and transcriptomic
variations in matched prefrontal cortex and cerebellum samples were identified
in apatient with a 47,XXY karyotype who was comorbid for SCZ and had a notably
reduced cerebellum mass compared with other individuals. Global DNA
methylation, assessed via the interrogation of LINE-1 and Alu repetitive
elements, was significantly altered in the 47, XXY patient in a tissue-specific
manner with extreme hypomethylation detected in the prefrontal cortex and
extreme hypermethylation in the cerebellum [115].
Fisher et al. [116] explored whether differences in DNA methylation at
age 10 were associated with monozygotic twin discordance for psychotic symptoms
at age 12. The Environmental Risk (E-Risk) Longitudinal Twin Study cohort of
2,232 children (1,116 twin pairs) was assessed for age-12 psychotic symptoms
and 24 monozygotic twin pairs discordant for symptoms were identified for
methylomic comparison. Site-specific DNA methylation differences were observed at
age 10 between monozygotic twins discordant for age-12 psychotic symptoms.
Similar DMPs were not found at age 5. The top-ranked psychosis-associated DMP (cg23933044), located in the promoter of
the C5ORF42 gene, was also
hypomethylated in post-mortem prefrontal cortex brain tissue from SCZ patients
compared to unaffected controls. Epigenetic variation in peripheral tissue is
associated with childhood psychotic symptoms and may indicate susceptibility to
SCZ and other mental health problems.
Epidemiological studies have identified prenatal exposure to famine as
a risk factor for SCZ. Analysis of gene expression and epigenetic modifications
in the brain of the offspring of the RLP50 rat, a recently developed animal
model of prenatal famine malnutrition exposure, indicate that offspring of
RLP50 exhibit differences in neurotransmitters and olfactory-associated gene
expression. In the hippocampus, the differentially-expressed genes are related
to synaptic function and transcription regulation. DNA methylome profiling of
the hippocampus also shows widespread but systematic epigenetic changes; in
most cases (87%) this involves hypermethylation. Genes encoded for the plasma
membrane are significantly enriched for changes in both gene expression and DNA
methylome profiling screens. Mecp2
and Slc2a1, two genes associated with
cognitive impairment, show significant down-regulation, and Slc2a1 is hypermethylated in the
hippocampus. Prenatal exposure to malnutrition leads to the reprogramming of
postnatal brain gene expression and epigenetic modifications contribute to the
reprogramming [117].
In the CNS, regulatory RNA networks and epigenetic mechanisms have
broad relevance to gene transcription changes involved in long-term memory
formation and cognition [118]. miR-137 is associated with SCZ and intellectual
disability. miR-137 acts as a potent player in regulating glutamatergic
synaptic transmission in the hippocampus by controlling the translation of
functionally critical genes at spatially opposite ends of the synapse,
contributing to the pathogenesis of cognitive impairments as seen in
neurodevelopmental disorders [119]. DISC-2,
Gomafu, EVF-2 and BDNF-AS are
lncRNAs associated with SCZ. These lncRNAs are responsible for specific
proteomic changes in SCZ [38].
Proteomic analysis indicates that SCZ and affective psychosis are
linked to a hypoglutamatergic state and hypofunction of energy metabolism,
while bipolar disorder and major depressive disorder (MDD) are linked to a
hyperglutamatergic state and hyperfunction of energy metabolism [120]. Proteins
with evidence for altered expression in SCZ are enriched for glutamate
signaling pathway proteins (GRIA4, GRIA3, ATP1A3, and GNAQ). Synaptic protein
co-expression is decreased in SCZ with the exception of a small group of postsynaptic
density proteins, whose co-expression increases and inversely correlates with
spine density in SCZ. Reduced ATP1A3 expression is supported by strong genetic
evidence indicating that it may contribute to psychosis and cognitive
impairment phenotypes [121].
Synapses are fundamental components of brain circuits and are disrupted
in over 100 neurological and psychiatric diseases. The synapse proteome is
physically organized into multiprotein complexes and polygenic mutations
converge on postsynaptic complexes in SCZ, autism and intellectual disability
[122]. The postsynaptic density (PSD) contains a complex set of proteins of
known relevance to neuropsychiatric disorders, and SCZ specifically.
Quantitative investigation of the PSD revealed more than 700 protein
identifications and 143 differentially expressed proteins. Prominent among
these were altered expression of proteins involved in clathrin-mediated
endocytosis (CME) (Dynamin-1, adaptor protein 2) and NMDA-interacting proteins
such as CYFIP2, SYNPO, SHANK3, ESYT and MAPK3. Pathway analysis of the
differentially expressed proteins implicated the cellular processes of
endocytosis, long-term potentiation and calcium signaling. Both single-gene and
gene-set enrichment analyses in genome-wide association data from the largest
SCZ sample to date of 13,689 cases and 18,226 controls show significant
association of HIST1H1E and MAPK3, and enrichment of this PSD proteome [123].
Methamphetamine produces a progressive increase in locomotor activity
(behavioral sensitization) in rodents that is believed to represent the
underlying neurochemical changes driving psychoses. Alterations to the
prefrontal cortex (PFC) are suggested to mediate the etiology and maintenance
of these behavioral changes. Proteomic analysis revealed 96 proteins that were
differentially expressed in the PFC of methamphetamine- treated rats, with 20%
of these being previously implicated in the neurobiology of SCZ in the PFC.
Proteins associated with synaptic regulation, protein phosphatase signaling,
mitochondrial function, and GABAergic network are disrupted in the PFC of SCZ
[124].
In serum proteomics of SCZ, over 140 proteins were found to be
different from other groups.Two protein peaks at the mass-to-charge ratio of
1,207.41 and 1,466.78 were markedly different, with the lowest expression in
specimens from SCZ patients. These proteins were identified as the N-terminal
fragments of fibrinogen [125]. Several markers (2-piperidinec carboxylic acid,
6-deoxy-mannofuranose, galactoseoxime and a serum peptide of m/z 3177) have an
attractive discriminating value in serum proteomics of SCZ [126]. The analysis
of whole saliva in SCZ revealed a 10-fold mean increase of α-defensins 1-4,
S100A12, cystatin A and S-derivatives of cystatin B levels, suggesting a
dysregulation of immune pathways in peripheral white blood cells [127].
The corpus callosum (CC), which is the largest portion of white matter
in the human brain and responsible for inter-hemispheric communication, is
altered in SCZ. CC proteomes were quantified by label-free spectral counting
and 5,678 unique peptides, corresponding to 1,636 proteins belonging to 1,512
protein families, were identified. Of those proteins, 65 differed significantly
in expression: 28 were upregulated and 37 downregulated. Among the
differentially expressed proteins are those associated with cell growth and
maintenance (neurofilaments and tubulins), cell communication and signaling
(14-3-3 proteins), and oligodendrocyte function (myelin basic protein,
myelin-oligodendrocyte glycoprotein) [128].
In a chronic phencyclidine (PCP) rat model in which glutamatergic
hypofunction is induced through noncompetitive NMDAR-receptor antagonism,
alterations in the levels of several cytokines (IL-5, IL-2, and IL-1β) and
fibroblast growth factor-2 were identified. Extensive proteomic and metabolomic
brain tissue profiling revealed a more prominent effect of chronic PCP
treatment on both the hippocampal proteome and metabolome compared to the
effect on the frontal cortex. Bioinformatic pathway analysis confirmed
prominent abnormalities in NMDA-receptor-associated pathways in both brain
regions, as well as alterations in other neurotransmitter systems such as
kainate, AMPA, and GABAergic signaling in the hippocampus and in proteins associated
with neurodegeneration [129].
Comparing disease and control cases, 58 unique differentially expressed
proteins were identified in SCZ, and 70 differentially expressed proteins in
bipolar disorder. Both disorders were characterized by alterations of proteins
involved in the oxidative stress response, mitochondrial function, and
protein-endocytosis, -trafficking, -degradation, and -ubiquitination focused on
GABAergic interneuron pathology in the hippocampus [130]. Human olfactory
neurosphere-derived (ONS) cells have been used to study the cellular pathology
of SCZ. Discovery-based proteomics and targeted functional analyses revealed
reductions in 17 ribosomal proteins, with an 18% decrease in the total
ribosomal signal intensity in SCZ-patient-derived ONS cells. Pathway analysis
of dysregulated proteomic and transcriptomic data sets from these ONS cells
converged to highlight perturbation of the eIF2α, eIF4 and mammalian target of
rapamycin (mTOR) translational control pathways, and these pathways were also
implicated in an independent induced pluripotent stem cell-derived neural stem
model, and cohort, of SCZ patients. Analysis in SCZ-genome-wide association
data from the Psychiatric Genetics Consortium specifically implicated eIF2α
regulatory kinase EIF2AK2, and confirmed the importance of the eIF2α, eIF4 and
mTOR translational control pathways at the level of the genome [131].
Depressive disorders
Recent evidence provides insights to epigenetic processes in
depression; however, replication is lacking and care must be taken in the
interpretation of current findings. Most studies have focused on DNA
methylation in various CNS or peripheral tissues, with almost universally small
sample sizes. Several epigenome-wide association studies have been reported and
the majority of studies have used a candidate-gene approach. Three genes (SLC6A4, BDNF, NR3C1) have been
investigated in more than one study [132]. Aberrant DNA methylation in the
blood of patients with major depressive disorder (MDD) has been reported in several
studies. Genome-wide DNA methylation profiling of peripheral leukocytes
detected diagnostic differences in DNA methylation at 363 CpG sites. All of
these loci showed greater DNA hypomethylation in patients with MDD than in
controls, and most of them (85.7%) were located in the CGIs in the gene
promoter regions [133]. A pilot study including an epigenome-wide methylation
analysis on the hippocampus and prefrontal cortex of depressive patients
revealed differential methylation profiles of 11 genes in hippocampus and 20
genes in prefrontal cortex, 5 of which were selected for replication of the
methylation status using pyrosequencing. Among these replicated targets, GRIN2A was found to be hypermethylated
in both prefrontal cortex and hippocampus. GRIN2A
encodes the glutamatergic N-methyl-D-aspartate receptor subunit epsilon-1
(NR2A) which is known to be involved in synaptic plasticity-related regulatory
processes probably disturbed in MDD [134]. Stress-induced maladaptive
transcriptional regulation in limbic neural circuits contributes to the
development of MDD, possibly through epigenetic factors that regulate chromatin
structure. Sun et al. [135] established that persistent upregulation of the ACF
(ATP-utilizing chromatin assembly and remodeling factor) ATP-dependent
chromatin-remodeling complex, occurring in the nucleus accumbens of
stress-susceptible mice and depressed humans, is necessary for stress-induced
depressive-like behaviors. Altered ACF binding after chronic stress correlates
with altered nucleosome positioning, particularly around the transcription
start sites of affected genes. These alterations in ACF binding and nucleosome
positioning are associated with repressed expression of genes implicated in
susceptibility to stress. The ACF chromatin-remodeling complex might be a
critical component in the development of susceptibility to depression and in
regulating stress-related behaviors.
The epigenetic regulation of BDNF may be involved in the
pathophysiology of MDD. As compared to healthy controls, MDD patients exhibit
reduced fractional anisotropy (FA) in the bilateral anterior and posterior
corona radiata (ACR and PCR), genu of the corpus callosum, and the bilateral
posterior thalamic radiations, and there is an inverse correlation between the
DNA methylation of the BDNF promoter
region and the FA of the right ACR in MDD patients. BDNF DNA methylation may contribute to structural white matter
changes in MDD patients [136]. Prenatal maternal psychological distress
increases risk for adverse infant outcomes. Prenatal depressive symptoms
significantly predict increased NR3C1 1F
DNA methylation in male infants and decreased BDNF IV DNA methylation in both male and female infants [137].
Buccal DNA hypermethylation at the two most widely studied BDNF promoters, I and IV, was associated with chronic late-life
depression. Three single-nucleotide polymorphisms (rs6265, rs7103411 and rs908867)
were also found to modify the association between depression and promoter I
methylation [138]. Intrapair DNA methylation differences in an intron of DEPDC7 (chr11:33040743) were associated
with intrapair differences in current depressive symptoms and in co-twin
studies [139]. Genome-wide methylome studies on depression suggest that, along
with differential DNA methylation, affected co-twins of monozygotic pairs have
increased DNA methylation variability, probably in line with theories of
epigenetic stochasticity. One differentially methylated probe (cg01122889) was located in the WDR26 gene, the DNA sequence of which
has been implicated in MDD. Expression of WDR26
has also been proposed as a biomarker of depression in human blood. Other genes
(CACNA1C, IGF2 and the p38 MAP kinase MAPK11)
showed differential variability [140].
Some neonates are affected by prenatal exposure to serotonin reuptake
inhibitor antidepressants (SRI) and maternal mood disturbances. Prenatal SRI
exposure was first associated with increased DNA methylation status primarily
at CYP2E1. Higher DNA methylation
status across 16 CpG sites and at each specific CpG site was associated with
exposure to lower 3rd trimester maternal depressed mood symptoms only in the
SRI-exposed neonates, indicating a maternal mood x SRI exposure interaction.
Higher DNA methylation levels at CpG2, CpG9 and CpG10, in the interrogated CYP2E1 region, were associated with
increased birth weight, independently of prenatal maternal mood, SRI drug
exposure, or gestational age at birth [141].
Greater DNA methylation in specific CpG sites at the serotonin
transporter promoter in peripheral cells is associated with childhood trauma,
depression, and smaller hippocampal volume [142]. There is an association of
fMRI blood oxygen level-dependent reactivity with the level of epigenetic
methylation of SLC6A4 in blood DNA in
patients with MDD. Activation in the anterior insula elicited by negative
emotional content was significantly positively associated with the degree of SLC6A4 methylation. Significantly
negative associations were observed between activation in the posterior insula
and the degree of SLC6A4 methylation
when judging the geometry of pictures after seeing negative, in contrast to
positive, emotional stimuli [143]. Imipramine, a major antidepressant, is known
to inhibit reuptake of serotonin and norepinephrine, which contributes to
recovery from MDD. Acute imipramine treatment inhibits NMDA receptor activity.
Acute imipramine treatment decreases Ca2+ influx through NMDA
receptors, whereas long-term treatment increases Ca2+ influx via the
same receptors. Long-term treatment increases NMDA receptor 2B (NR2B) subunit expression via epigenetic
changes, including increased acetylation of histones H3K9 and H3K27 in the NR2B
promoter and decreased activity of histone deacetylase 3 (HDAC3) and HDAC4
[144]. Treatment with venlafaxine decreases expression of prolyl 4-hydroxylase
(P4HB), ubiquitin-conjugating enzyme E2K (HIP2) and plastin 3 (T-plastin), and
up-regulates expression of growth factor beta-3 (TGF-β3), dihydropyrimidinase-like 3
(DPYSL3), and pyruvate kinase (PKM) after differentiation for 1 and 7 days
[145].
Disturbances of the hypothalamic-pituitary-adrenal axis have been
implicated in the pathophysiology of bipolar disorder and MDD. BD patients have
significantly increased levels of the major pituitary hormones
pro-opiomelanocortin (POMC) and galanin. Bipolar patients also show changes in
proteins associated with gene transcription, stress response, lipid metabolism,
and growth signaling. In contrast, MDD patients have significantly decreased
levels of the prohormone-converting enzyme carboxypeptidase E and follow-up
enzymatic analysis showed decreased activity of prolyl-oligopeptidase
convertase. Altered prohormone processing may occur in pituitaries of MDD
patients. MDD patients also have significant changes in proteins involved in
intracellular transport and cytoskeletal signaling [146].
Quantitative proteomic studies identified 10 proteins that were
consistently upregulated or downregulated in MDD patients. 3 proteins
(ceruloplasmin, inter-alpha-trypsin inhibitor heavy chain H4 and complement
component 1qC) were upregulated during the depressive status [147]. Combined
proteomic and metabolomic approaches may provide a comprehensive understanding
of MDD's etiology and contribute to the identification of diagnostic
biomarkers. The combined analyses found significant alterations associated with
cerebellar energy metabolism in animal models, including (i) abnormal amino acid
metabolism accompanied by corresponding metabolic enzymatic alterations and
disturbed protein turnover, (ii) increased glycolytic and tricarboxylic acid
(TCA) cycle enzyme levels paralleled by changes in the concentrations of
associated metabolites, and (iii) perturbation of ATP biosynthesis through
adenosine accompanied by perturbation of the mitochondrial respiratory chain
[148].The differential proteomic analysis of urine samples from first-episode
drug-naïve MDD subjects and healthy controls (HC) identified a total of 27
differential proteins, primarily including enzymes, plasma proteins, serpins,
and adhesion molecules. The arginine recycling enzymeargininosuccinate synthase
(ASS1) was confirmed to be significantly downregulated in the urine of 30 depressed
subjects while remaining unchanged in plasma [149].
Post-stroke depression (PSD) is the most common psychiatric
complication facing stroke survivors and has been associated with increased
distress, physical disability, poor rehabilitation, and suicidal ideation.
Plasma proteomics identified 6
proteins associated with lipid metabolism and immunoregulation (apolipoprotein
A-IV (ApoA-IV), apolipoprotein C-II (ApoC-II), C-reactive protein (CRP),
gelsolin, haptoglobin, and leucine-rich alpha-2-glycoprotein (LRG)). ApoA-IV
expression was significantly upregulated in PSD as compared to stroke subjects.
ApoC-II, LRG, and CRP expression were significantly downregulated in both PSD
and HC subjects relative to stroke subjects. Gelsolin and haptoglobin expression
were significantly dysregulated across all three groups [150].
Other psychiatric
disorders
Many other psychiatric disorders exhibit epigenetic anomalies including
drug abuse and addictive behaviors [151-153], alcohol spectrum disorders [154],
sexual disorders [155], and post-traumatic stress disorder [156-158]. Prenatal
alcohol exposure (PAE) can cause fetal alcohol spectrum disorders (FASD).
Children born with FASD have unique DNA methylation defects that can be
influenced by sex and medication exposure [159]. Altered DNA methylation at the
aryl hydrocarbon receptor repressor (AHRR) correlates with self-reported
smoking. Smoking was associated with DNA demethylation at two distinct
loci within AHRR (cg05575921 and cg21161138), and methylation status at
the AHRR residue interrogated by cg05575921
was highly correlated with serum cotinine levels [72].
Neurodegenerative
Disorders
Epigenetic dysregulation is an attractive mechanism to explain in part
enigmatic areas of confusion associated with the pathogenesis of age-related
neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease,
and Huntington's disease (Table 1),
where it may mediate interactions between genetic and environmental risk
factors, or directly interact with disease-specific pathological factors [160].
Alzheimer’s disease
Alzheimer’s disease (AD) is the most frequent neurodegenerative
disorder in the elderly population. Over 600 different genes distributed across
the human genome are potentially involved in AD pathogenesis, where
environmental factors and epigenomic aberrations also participate [14,161-164].
Conventional genomics do not explain in full AD pathogenesis in which
epigenetics may help to understand some obscure events. Major epigenetic events
may contribute to AD pathology, although evidence is still very limited
[17,165-167]. Pharmaceuticals, pesticides, air pollutants, industrial
chemicals, heavy metals, hormones, nutrition, and behavior can change gene
expression through a broad array of gene regulatory mechanisms (gene
translocation, histone modifications, DNA methylation, DNA repair,
transcription, RNA stability, alternative RNA splicing, protein degradation,
gene copy number, and transposon activation) [168]. Genetic variation
associated with different diseases interferes with microRNA-mediated regulation
by creating, destroying, or modifying miRNA binding sites. miRNA-target
variability is a ubiquitous phenomenon in the adult human brain, which may
influence gene expression in physiological and pathological conditions. One of
the major roles of lncRNAs in the nucleus is the regulation of gene expression
at the transcriptional level via histone or DNA modification [169]. Epigenetic
mechanisms and miRNAs have recently been shown to closely interact with each other,
thereby creating reciprocal regulatory circuits, which appear to be disrupted
in AD [74]. Brain hypoperfusion-related changes in DNA methylation may also
contribute to accelerate neuronal death. Short-term, sub-lethal hypoxia results
in long-lasting changes to genome-wide DNA methylation status and some of these
changes can be highly correlated with transcriptional modulation in a number of
genes involved in functional pathways [170].
Inflammatory mechanisms contribute substantially to secondary tissue injury
after brain ischemia. Regulatory T cells (RTC) are endogenous modulators of
postischemic neuroinflammation. HDACi, using trichostatin A, increases the
number of RTC, boosts their immunosuppressive capacity and interleukin (IL)-10
expression, reduces infarct volumes and behavioral deficits after cortical
brain ischemia, attenuates cerebral proinflammatory cytokine expression, and
increases the number of brain-invading RTC. A similar effect is obtained using
tubastatin, a specific inhibitor of HDAC6 and a key HDAC in Foxp3 regulation. The neuroprotective
effect of HDACi depends on the presence of Foxp3+
RTC, and in vivo and in vitro studies show that the anti-inflammatory cytokine
IL-10 was their main mediator [171].
Memory decline is a seminal symptom in dementia. Gene expression is
required for long-lasting forms of memory. Epigenetic mechanisms do not only
provide complexity in the protein regulatory complexes that control coordinate
transcription for specific cell function, but the epigenome encodes critical
information that integrates experience and cellular history for specific cell
functions as well. Epigenetic mechanisms provide a unique mechanism of gene
expression regulation for memory processes. Negative regulators of gene
expression, such as HDACs, have powerful effects on the formation and
persistence of memory. HDAC inhibition transforms a subthreshold learning event
into robust long-term memory and generates a form of long-term memory that
persists beyond the point at which normal long-term memory fails [172]. Whereas
increments in histone acetylation have consistently been shown to favor
learning and memory, a lack thereof has been causally implicated in cognitive
impairments in neurodevelopmental disorders, neurodegeneration and aging. As histone
acetylation and cognitive functions can be pharmacologically restored by
histone deacetylase inhibitors, this epigenetic modification might constitute a
molecular memory aid on the chromatin and, by extension, a new template for
therapeutic interventions against cognitive decline [27].
Neurons, due to their post-mitotic state, high metabolism, and
longevity are particularly prone to the accumulation of DNA lesions. DNA damage
has been suggested as a major contributor to both age-associated neurodegenerative
diseases and acute neurological injury. The DNA damage response is a key factor
in maintaining genome integrity. It relies on highly dynamic post-translational
modifications of the chromatin and DNA repair proteins to allow signaling,
access, and repair of the lesion [173]. The repair of DNA lesions, particularly
oxidative DNA lesions, might be altered in AD. DNA damage is paralleled by a
decrease in DNA repair activities. DNA repair proteins might be inactivated by
oxidative induced post-translational modifications or degradation. Activation
of DNA repair pathways might generate death signals ending with neuronal
apoptosis. A link between environmentally-induced epigenetic modification,
oxidation, and repair of AD-related genes has been proposed [174]. Early life
exposure of rodents and primates to xenobiotics may enhance the expression of
genes associated with AD, repress the expression of others, and increase the
burden of oxidative DNA damage in the aged brain. Epigenetic mechanisms that
control gene expression and promote the accumulation of oxidative DNA damage
are mediated through alterations in the methylation or oxidation of CpG
dinucleotides. Environmental influences occurring during brain development
inhibit DNA-methyltransferases, thus hypomethylating promoters of genes
associated with AD, such as the APP.
This early life imprint may persist and be triggered later in life to increase
the levels of APP and Aβ.
Increased Aβ levels promote
the production of reactive oxygen species, which damage DNA and accelerate
neurodegenerative events. These early life perturbations may result in
hypomethylation as well as hypermethylation of genes. The hypermethylated genes
are rendered susceptible to Aβ-enhanced oxidative DNA damage because methylcytosines restrict repair
of adjacent hydroxyguanosines [175].
Studies performed in brains and peripheral tissues of both AD patients
and individuals affected by mild cognitive impairment (MCI) revealed that
oxidative DNA damage is one of the earliest detectable events during the
progression from healthy aging to dementia. Some authors have suggested that
mutations or polymorphisms in DNA repair genes might impair DNA repair.
However, this hypothesis does not seem to be confirmed by recent genetic
association studies. The growth arrest and DNA damage-inducible (Gadd) 45
proteins have been associated with numerous cellular mechanisms including
cell-cycle control, DNA damage sensation and repair, genotoxic stress,
neoplasia, and molecular epigenetics. Gadd45-related
genes have been implicated in a host of normal and aberrant CNS processes,
including early and postnatal development, injury, cancer, memory, aging,
psychiatric disease, and neurodegenerative disorders. The proteins act through
a variety of molecular signaling cascades including the MAPK cascade,
cell-cycle control mechanisms, histone regulation, and epigenetic DNA
demethylation [176].
Brain aging and AD are associated with epigenetic dysregulation at
various levels [177]. Twin studies in AD support the notion that epigenetic
mechanisms mediate the risk for AD.However, it is still not fully clear whether
the observed epigenetic changes actually represent a cause or a consequence of
the disease [23].
DNA Methylation of
pathogenic genes: Many
AD-related genes contain methylated CpG sites in their promoter regions, and a
genome-wide decrease in DNA methylation has been reported in AD [17,166].
Methylation status of repetitive elements (i.e. Alu, LINE-1 and SAT-α) is a major contributor of global DNA
methylation patterns.The study of global DNA methylation levels for long
interspersed nuclear element 1 (LINE-1) repetitive sequences in patients with
AD and controls did not provide clear results. In one study, no differences in LINE-1 methylation levels between
patients and controls were found [178] whereas in another study LINE-1 methylation was found increased
in AD patients compared with healthy volunteers [179]. In AD, both
hypomethylation and hypermethylation of specific genes have been reported [17].
DNA methylation of the APP promoter
was found to be decreased in the brain of autopsy cases older than 70 years of
age as compared with younger cases [180]. The intracellular domain of APP (AICD)
has emerged as a key epigenetic regulator of gene expression controlling a
diverse range of genes, including APP
itself, the amyloid-degrading enzyme neprilysin,
and aquaporin-1 [181]. Abnormal
processing of neuronal cell membrane APP is accompanied by elevated human serum
and CSF levels of 24-hydroxycholesterol, an endogenous ligand of Liver X
receptor (LXR-α). There is an
epigenomic pathway that connects LXR-α activation with genes involved in the regulation of aberrant Aβ production leading to the generation
of toxic and inflammatory mediators responsible for neuronal death. LXR-α activation by its specific
endogenous or exogenous ligands results in the overexpression of the PAR-4 gene and suppression of the AATF gene through its inherent capacity
to regulate genes coding for SREBP and NF-κB. Overexpression of the PAR-4 gene is accompanied by aberrant Aβ production followed by ROS
generation and subsequent neuronal death. Aβ-induced heme oxygenase-1 can
ensure cholesterol-oxidation to provide endogenous ligands for the sustained
activation of neuronal LXR-α-dependent
epigenomic pathways leading to neuronal death in AD [182].
Presenilin1 (PSEN1) is
modulated by DNA methylation in neuroblastoma cells and Alzheimer's mice in an
experimental model of nutritionally altered one-carbon metabolism. Studies
performed on human neuronal cell cultures revealed that folate and other
B-vitamins deprivation from the media resulted in epigenetic modification of
the PSEN1 gene [183].
Several pathogenic genes (APP,
PS1, APOE, BACE) and many other AD-related susceptibility genes contain
methylated CpG sites. The promoter region of the APP gene is hypomethylated, this contibuting to a potential
enhancement of Aβ
production; however, some authors have reported non-relevant changes in APP
methylation, with an epigenetic drift in AD samples [184]. BACE and PS1expression is
enhanced after folate deprivation-induced hypomethylation and restored when
folate deficiency is supplemented with SAM. Aβ may induce genome-wide
hypomethylation accompanied by upregulation of genes involved in
neuroinflammation (TNF) and apoptosis
(caspase-3), which contribute to Aβ production, the process thus
entering into a vicious circle [17].
The APOE gene exhibits a bimodal
structure, with a hypomethylated CpG-poor promoter and a fully methylated
3’-CpG-island, containing the sequences for the APOE4-haplotype. According to Wang et al. [17,184], aberrant
epigenetic changes in this CpG-island may contribute to LOAD pathology. A
hypermethylated CpG-island is present within the APOE gene. The APOE4 sequence
may change the epigenetic function of the methylated 3’-CpG islands in the APOE4 allele by the C to T transition
that is involved in a loss of a methylatable CpG unit [184]. APOE4 carriers show a dose-dependent
risk, and the relative mRNA level of APOE4
is increased in AD compared to controls, indicating that variability in the
neuronal expression of APOE contributes
to disease risk [185].
Clusterin gene (CLU)
(apolipoproteinJ, ApoJ), together with APOE,
influence both Aβ
aggregation and clearance. CLU levels
are increased in AD and may be associated with brain atrophy, disease severity,
and clinical progression. The promoter region of CLU contains a CpG-rich methylation domain. The demethylating
effect of 5-aza-2’ deoxycytidine in prostate cancer cell lines increases the
expression of CLU [186].
Hyperphosphorylated tau is responsible for the formation of NFTs.
Changes in methylation status differ among transcription factor binding sites
of tau promoter. Binding sites for GCF (granulocyte chemotactic factor),
responsible for repression of GC-rich promoters, were found to be
hypomethylated, whereas binding sites for the transcriptional activator SP1 (specificity factor 1) were
hypermethylated [187]. High levels of Hcy may induce tau hyperphosphorylation,
NFT formation, and SP formation via inhibition of methyltransferases and
hypomethylation of protein phosphatase 2A (PP2A), a dephosphorylating enzyme of
phosphorylated tau [188]. In transgenic APPswe/presenilin (PS) 1 (A246E) mice, PP2A methylation at L309 site is
decreased, in parallel with increased tau phosphorylation at Tau-1 and
PHF-1sites. Aβ25-35 induces
demethylation and enhances tau phosphorylation [189]. Hypomethylation of PP2A may lead to tau
hyperphosphorylation and NFT formation [17].
Sánchez-Mut et al. [190] studied 12 distinct mouse brain regions
according to their CpG 5'-end gene methylation patterns, and the DNA methylomes
obtained from the cerebral cortex were used to identify aberrant DNA
methylation changes that occurred in two mouse models of AD. They translated
these findings to patients with AD and identified DNA methylation-associated
silencing of three target genes: thromboxane A2 receptor (TBXA2R), sorbin and SH3 domain containing 3 (SORBS3), and spectrin beta 4 (SPTBN4).
These hypermethylation targets suggest that the cyclic AMP response
element-binding protein (CREB) activation pathway and the axon initial segment
might contribute to AD pathology.
Several components of the cell cycle (P16, P21, P27, P53, RB1,
cyclinB2, alternate open reading frame (ARF) protein product) and apoptosis
pathways (caspase1, 3, 7, 8, 9) are regulated by DNA methylation and appear
upregulated in AD neurons. SORBS3 (vinexin,
SCAM-1 or SH3D4), encoding a cell
adhesion molecule expressed in neurons and glia, is progressively
hypermethylated with age. S100A2, a
member of the S100 family of calcium binding proteins, which exhibits an
age-dependent decrease in DNA methylation later in life, is also
hypermethylated in AD [17].
Chaperones may have a crucial role in AD due to their involvement in
protein quality control, folding, and degradation. Silva et al. [191]
investigated the mRNA and promoter DNA methylation levels of two chaperones, HSPA8 and HSPA9, in postmortem brain tissue (entorhinal and auditory cortices
and hippocampus) from healthy elderly and AD subjects as well as in peripheral
blood of healthy elderly and AD patients. No changes were observed in
peripheral HSPA8 and HSPA9 expression between elderly controls and AD. A
significant downregulation of HSPA8 and HSPA9 was observed in AD across the
three brain regions compared to the controls.
Summarizing, DNA methylation changes are present in AD-related genes;
some of these genes are hypermethylated (MTHFR,
Neprilysin, MAPT, APOE, SORB3), while others have been found to be
hypomethylated (APP, BACE, PSEN1, PP2A,
S100A2, CREB5) [17,23]. DNA methylation of CpG units by DNMTs disrupts the
binding of transcription factors and attracts methyl-CpG-binding domain
proteins that are associated with gene silencing and chromatin compaction
[192].
Histone
modifications: A
small bulk of recent information [17,27,193] suggests that histone
modifications are present in AD: (i) histone acetylation is reduced in AD brain
tissues [194] and in AD transgenic models [27]; (ii) levels of HDAC6, a
tau-interacting protein and a potential modulator of tau phosphorylation and
accumulation, are increased in cortical and hippocampal regions in AD [195]; mice
lacking HDAC6 are cognitively normal but reducing endogenous HDAC6 levels
restores learning and memory and α-tubulin acetylation [196]; (iii) SIRT1 is decreased in the parietal
cortex of AD patients, and the accumulation of Aβ and tau in AD brains might be
related to the loss of SIRT1 [197], since SIRT1 may reduce Aβ production activating the
transcription of ADAM10 [198]; (iv)
in the brains of twins discordant for AD, trimethylation of H3K9, a marker of
gene silencing, and condensation of heterochromatin structure, are increased in
the temporal cortex and hippocampus of the AD twin as compared to the twin
devoid of AD neuropathology [199]; (v) phosphorylation of H3S10, a key
regulator in chromatin compaction during cell division, is increased in the cytoplasm
of hippocampal neurons in AD cases [200]; (vi) evidence of DNA damage, as
reflected by phosphorylated H2AX at Ser139, is present in hippocampal
astrocytes of AD patients [201]; (vii) long-term potentiation (LTP) and memory
deficits in APP/PS1 transgenic mice might be mediated in part by decreased H4
acetylation; improving histone acetylation level restores learning after
synaptic dysfunction [202]; (viii) acetylation of H3 and H4 is increased in
3xTg-AD neurons relative to non-transgenic neurons [203]; (ix) nuclear
translocation of EP300 interacting inhibitor of differentiation 1 (EID1), aCBP/p300 inhibitory protein, is increased in the cortical neurons
of AD patients, and overexpression of EID1
is reported to reduce hippocampal LTP and to impair cognitive function by
inhibiting CBP/p300 acetyltrasferase activity and disrupting neuronal structure
[204]; (x) memory formation leads to a transient increase of acetylation only
in residues within H2B, H3, H4 [205,206]; (xi) HDAC inhibition induces
dendritic sprouting, increases synaptic number, and improves long-term memory
[207]; (xii) overexpression of neuronal HDAC2 decreases dendritic spine
density, synapse number, synaptic plasticity and memory formation, whereas
HDAC2 deficiency increases synapse number and memory facilitation [208,209];
(xiii) HDAC4 is involved in learning and synaptic plasticity, and selective
inhibition of HDAC4 activity may deteriorate learning and memory [210]; (xiv)
treatment of hippocampal neurons with HDAC inhibitors facilitates Bdnf expression via hyperacetylation of
histones at the Bdnf promoters
[211,212]; (xv) histone (H3K4) methylation participates in the regulation of Bdnf expression and memory formation
[213]; (xvi) histone methylation also facilitates memory consolidation coupled
with histone acetylation; inhibition of HDACs with sodium butyrate (NaB) causes
an increase in H3K4 trimethylation and a decrease in H3K9 dimethylation in the
hippocampus after fear conditioning
[213]; (xvii) histone H3 acetylation, methylation and phosphorylation are
increased in the prefrontal cortex of Tg2576 mice, and histone H4 acetylation
is increased in the hippocampal CA1 neurons of these transgenic mice [214].
Age-related differences in epigenetic acetylation and methylation of
histones are associated with age-related gene regulation. In studies to
quantify single cell acetylation and methylation levels across the life span in
cultured hippocampal/cortical neurons from the 3xTg-AD mouse model and from
non-transgenic mice, Walker et al. [203] found that in non-transgenic neurons,
H3 acetylation was unchanged with age, while H4 acetylation decreased with age
of the donor. Compared to non-transgenic neurons, 3xTg-AD neurons had higher levels
of H3 and H4 acetylation beginning at 4 months of age. In contrast to
non-transgenic neurons, 3xTg-AD neurons increased acetylation with age; 3xTg-AD
neurons also responded differently to inhibition of histone deacetylases at an
early age. Treatment of non-transgenic neurons with the AD peptide Aβ also elevated levels of
acetylation. The repressive function of histone H3 lysine 9 (H3K9)-methylation
increased with age in non-transgenic neurons, which was amplified further in
3xTg-AD neurons. The dominant effect of higher H3K9 methylation was supported
by lower Bdnf gene expression in
non-transgenic and 3xTg-AD mice. The epigenetic states of non-transgenic and
3xTg-AD brain neurons are profoundly different and reversible, beginning at 4
months of age when the first memory deficits are reported [203].
Nucleosome remodeling is carried out by chromatin remodeling complexes
(CRCs) that interact with DNA and histones to physically alter chromatin
structure and ultimately regulate gene expression. Human exome sequencing and
genome-wide association studies have linked mutations in CRC subunits to
intellectual disability disorders, autism spectrum disorder and schizophrenia.
There appear to be both developmental- and adult-specific roles for the
neuron-specific CRC nBAF (neuronal Brg1/hBrm
Associated Factor). nBAF regulates
gene expression required for dendritic arborization during development, and in
the adult, contributes to long-term potentiation, a form of synaptic
plasticity, and long-term memory. Vogel-Ciernia and Wood [215] proposed that
the nBAF complex is a novel
epigenetic mechanism for regulating transcription required for long-lasting
forms of synaptic plasticity and memory processes and that impaired nBAF
function may result in human cognitive disorders.
Histone deacetylase 6 (HDAC6) expression increases significantly in the
hippocampus and other relevant brain regions in both patients with AD and
animal models of AD. However, when and how HDAC6 expression increases during
the course of AD progression remains unclear. Increased HDAC6 expression
contributes to AD-associated neurodegeneration, although beneficial effects
have also been identified in some pathogenic mechanisms (axonal growth and
transport, synaptic plasticity, oxidative stress, apoptosis, neuroinflammation,
misfolded proteins and aggregates) [216].
Sleep disruption associated with AD is driven by epigenetic changes
mediated by the histone acetyltransferase (HAT) Tip60. Tip60 functionally
interacts with the AD-associated amyloid precursor protein (APP) to regulate
axonal growth of Drosophila small ventrolateral neuronal (sLNv) pacemaker
cells, and their production of neuropeptide pigment dispersing factor (PDF)
that stabilizes appropriate sleep-wake patterns in the fly. Loss of Tip60 HAT
activity under APP neurodegenerative conditions causes decreased PDF
production, retraction of the sLNv synaptic arbor required for PDF release and
disruption of sleep-wake cycles in these flies. Excess Tip60 in conjunction
with APP fully rescues these sleep-wake disturbances by inducing
overelaboration of the sLNv synaptic terminals and increasing PDF levels,
supporting a neuroprotective role for Tip60 in these processes [217].
The sirtuins are NAD+-dependent histone/protein deacetylases
that are similar to Saccharomyces
cerevisiae silent information regulator 2 (Sir2). Sirtuins regulate various
normal and abnormal cellular and metabolic processes, including tumorigenesis,
neurodegeneration, and processes associated with type 2 diabetes and obesity.
Several age-related diseases, such as AD, and longevity have also been linked
to the functions of sirtuins [218].
Chromatin modification is an important epigenetic mechanism underlying
neuroplasticity. A chromatin-modifying complex, containing the histone
demethylase PHF8 and the acetyltransferase TIP60, is a key regulator of the
activity-induced expression of Arc, a mediator of synaptic plasticity.
Mutations in PHF8 cause X-linked mental retardation while TIP60 has been
associated with AD. With synaptic activity, this dual function complex is
recruited to the Arc promoter, where it specifically counteracts the
transcriptionally repressive histone mark H3K9me2 to facilitate the formation
of the transcriptionally permissive H3K9acS10P, thereby favoring
transcriptional activation. Gain-of-function of the PHF8-TIP60 complex in
primary rat hippocampal neurons has a positive effect on early activity-induced
Arc gene expression, whereas interfering with the function of this complex
abrogates it. The majority of common interactors of PHF8 and TIP60 are involved
in mRNA processing, including PSF, an important molecule involved in neuronal
gene regulation. PHF8 and TIP60 interact at the single molecule level with PSF,
situating this chromatin-modifying complex at the crossroads of transcriptional
activation. These data reported by Oey et al. [219] indicate that an epigenetic
pathway can regulate neuronal activity-dependent gene transcription.
Non-coding RNAs:Several lncRNAs are dysregulated
in AD (Sox2OT, 1810014B01Rik, BC200, BACE1-AS, NAT-Rad18, 17A, GDNFOS),
Parkinson’s disease (naPINK1, Sox2OT, 1810014B01Rik, BC200), and Huntington’s
disease (HAR1F, HTTAS, DGCR5, NEAT1, TUG1) [33]. miRNAs belong to the
class of non-coding regulatory RNA molecules of ∼22 nt length and are
now recognized to regulate ∼60% of all known genes through
post-transcriptional gene silencing (RNA interference) (RNAi). Alterations in
epigentically regulated miRNAs may contribute to the abnormal expression of
pathogenic genes in AD [33,74]. Examples
of miRNAs directly linked to AD pathogenesis include miR-34a (1p36.22),
miR-34b/c (11q23.1), miR-107 (10q23.31),miR-124 (8p23.1/8p12.3/20q13.33),
miR-125b (11q24.1/21q21.1), and miR-137 (1p21.3); and examples of
epigenetically regulated miRNAs with targets linked to AD pathogenesis are
let-7b (22q13.1), miR-9 (1q22/5q14.3/15q26.1), miR-132/212 (17p13.3), miR-146a
(5q34), miR-148a (7p15.2),miR-184 (15q25.1), and miR-200 (miR-200b/200a/429,
1p36.33; miR-200c/141, 12p13.31) [74].
miRNAs can be used as biomarkers to discriminate
different disease forms, staging and progression, as well as prognosis [220]. A unique
circulating 7-miRNA signature (hsa-let-7d-5p, hsa-let-7g-5p, hsa-miR-15b-5p,
hsa-miR-142-3p, hsa-miR-191-5p, hsa-miR-301a-3p and hsa-miR-545-3p) reported by
Kumar et al. [220] in
plasma, could distinguish AD patients from normal controls with >95%
accuracy. Leidinger
et al.[221] showed a novel miRNA-based signature for detecting AD from blood
samples. Using this 12-miRNA signature, they differentiated between AD and
controls with an accuracy of 93%, a specificity of 95% and a sensitivity of
92%. The differentiation of AD from other neurological diseases (MCI, multiple
sclerosis, Parkinson disease, major depression, bipolar disorder and
schizophrenia) was possible with accuracies between 74% and 78%. Alexandrov et
al. [222] found
increased levels of miRNA-9, miRNA-125b, miRNA-146a, and miRNA-155 in the CSF
and brain tissue-derived extracellular fluid from patients with AD, suggesting
that these miRNAs might be involved in the modulation or proliferation of
miRNA-triggered pathogenic signaling in AD brains.
AD-related SNPs interfere with miRNA gene regulation and
affect AD susceptibility. The significant interactions include target SNPs
present in seven genes related to AD prognosis with the miRNAs- miR-214, -23a
& -23b, -486-3p, -30e*, -143, -128, -27a &-27b, -324-5p and -422a. The
dysregulated miRNA network contributes to the aberrant gene expression in AD
[223-225].
Several miRNAs have been identified in vitro to
directly regulate the APP mRNA,
including miRNA let-7, the miR-20a family (miRs-20a, -17 and -106b), miRs-106a
and 520c, miR-101, miR-16, and miRs-147, -153, -323-3p, -644 and -655 [17]. Inhibition of
miR-101 overexpression reduces APP and Aβ load in hippocampal neurons [226]. MiR-16 targets APP
to potentially modulate AD pathogenesis, and miR-16 overexpression may lead to
reduced APP expression [227]. Both miR-124 and
polypyrimidine tract binding protein1 (PTBP1) may alter splicing of APP exons 7 and 8 in neuronal cells [228]. miR-124 also
regulates the expression of BACE1 [229]. mRNA expression of BACE1 is mediated by both miRNAs
(miRs-9, -29a/b-1, -29c, -107, -298, -328 and-485-5p) and long ncRNAs (BACE1-antisense) (BACE1-AS), and is repressed by miRs-29a, -29b-1 and -9 in vitro. In
transgenic HEK293-APP cells, transient miR-29a/b-1ower expression decreases
BACE1 levels and Aβ production [230]. miR-29 overexpression lowers BACE1 protein levels [231]. miRNAs repress BACE1 through direct binding to
sequences in its 3’ untranslated region (3’UTR), whereas miR-485-5 prepresses BACE1 via binding to its open reading
frame in exon 6. miR-107 is downregulated at intermediate stages (Braak
stage-3) of AD pathogenesis and might accelerate AD progression through control
of BACE1 [232]. miR-298, miR-328
and miR-195 inversely correlate with BACE1 protein, and downregulate Aβ levels
by inhibiting the translation of BACE1[233,234]. miR-125 decreases
whereas BACE1 increases in animal
models [234].
Overexpression of miR-485-5p reduces BACE1 protein levels by 30% while
knockdown of miR-485-5p increases BACE1 protein levels [235]. BACE1-AS, a∼2kb conserved ncRNA
transcribed from the opposite strand to BACE1
and co-expressed with BACE, is up-regulated in AD, potentially promoting Aβ
generation and AD pathogenesis. BACE1-AS may
enhance BACE1 mRNA stability by
“masking” the binding site for miR-485-5p and preventing miRNA-induced
translational repression of BACE1 mRNA
[235,236].
The RNA polymerase III-dependent ncRNA, NDM29,
promotes APP amyloidogenesis and Aβ secretion [237]. miR-107 levels are
reduced in AD temporal cortex [238,239]. Loss of miRs-9,29a/b-1, -137and -181c (currently
down-regulated in AD frontal cortex) increases Aβ production and serine
palmitoyltransferase (SPT), the first rate-limiting enzyme in ceramide
biosynthesis [240].
miRNA-106b (down-regulated in anterior temporal cortex) can influence Aβ
metabolism either through direct regulation of APP itself, or via modulating
APP trafficking, Aβ clearance and β- and γ-secretase activity through
regulation of the ATP-binding cassette transporter A1 (ABCA1), which is
elevated in the hippocampus, correlating with cognitive decline [241]. The brain-expressed
ncRNA, 17A, is up-regulated in the AD cortex, promoting Aβ in response to
neuroinflammation injury [242].
Several miRNAs also regulate tau metabolism.
The miR-132/PTBP2 pathway influences MAPT
exon 10 splicing in brain and may contribute to AD pathogenesis. miR-132
was found to be down-regulated in some tauopathies, such as progressive
supranuclear palsy (PSP), a major 4R-tau tauopathy, where the protein levels of
the neuronal splicing factor PTBP2 were elevated [243]. miR-124, -9, -132
and -137 might regulate the 4R/3R ratio in neuronal cells [243]. Both miR-9 and
miR-124 are down-regulated in AD and might affect tau. The miR-15/ERK1 pathway
mediates tau phosphorylation. miR-15a is down-regulated in AD brains [244]. The miR-15 family
(miR-15a, -16, -195 and -497) targets extracellular signal-regulated kinase
1(ERK1) expression; and decreased miR-15 levels might participate in neuronal
tau hyperphosphorylation. miR-26a represses mRNA of the tau kinase GSK-3β
involved in Aβ production and NFT formation [245,246]. miR-26a expression
is also altered in AD [247]. In
conditional Dicer knockout mice, with reduced brain miRNA production, tau
hyperphosphorylation and altered MAPT
splicing is observed; and reduced miRNA processing in dicer-1 knockout flies
enhances tau-induced neurodegeneration [248].
SIRT1 deacetylates tau and SIRT1 deficiency
increases tau acetylation and the accumulation of hyperphosphorylated tau [197,249]. miR-9, -34c
and-181c repress SIRT1 mRNA [250,251]. miR-128 modulates
the expression of BAG2, the cochaperone involved in tau degradation and
aggregation [252]. miR-212
is down-regulated in AD, and appears to be involved in NFT density [238,239]. miR-146a is an
inflammation effector associated with immune and inflammation signaling by
targeting IRAK1. miR-146a upregulation in AD brain might contribute to
neuroinflammation [253,254]. miR-146a
interacts with the 3’UTR of Complement factor H (CFH), a repressor of the
inflammatory response, which is down-regulated in AD [255]. miRNA-146a is an inducible,
22-nucleotide, small RNA over-expressed in AD brain. Up-regulated miRNA-146a
targets several inflammation-related and membrane-associated messenger RNAs
(mRNAs), including those encoding complement factor-H (CFH) and the
interleukin-1 receptor associated kinase-1 (IRAK-1), resulting in significant
decreases in their expression. The most significant miRNA-146a-CFH changes are
found in HMG cells, the 'resident scavenging macrophages' of the brain [256]. miR-101
interacts with cyclooxygenase-2 (COX-2), and downregulation of miR-101 might
induce COX-2 upregulation in AD,
enhancing the inflammatory response [226]. miR-124, -125b, -132, -134, -138 and -219 influence
synaptic plasticity. miR-132 is down-regulated and miR-125bis up-regulated in
different AD brain regions, probably affecting miniature excitatory
postsynaptic currents (mEPSCs) [257].
The INK4b-ARF-INK4a locus
encodes for two cyclin-dependent kinase inhibitors, p15 (INK4b) and p16 (INK4a)
and a regulator of the p53 pathway, ARF. ANRIL, a non-coding RNA, is also
transcribed from the locus. ARF, p15 (INK4b), and p16 (INK4a) are
well-established tumor suppressors whose function is frequently disabled in
human cancers. SNPs mapping in the vicinity of ANRIL are linked to a wide spectrum of conditions, including
cardiovascular disease, ischemic stroke, type 2 diabetes, frailty and AD. The INK4b-ARF-INK4a locus is regulated by
Polycomb repressive complexes (PRCs), and its expression can be invoked by
activating signals. Other epigenetic modifiers, such as the histone
demethylases JMJD3 and JHDM1B, the SWI/SNF chromatin remodeling complex, and
DNMTs regulate the locus interplaying with PRCs [258].
Proteomics: There is a great interest in
developing specific, sensitive, and practical tools to differentially diagnose
and discriminate the different types of dementia. The currently available
cerebrospinal fluid (CSF) biomarkers for AD (Aβ, total Tau (t-Tau),
phosphorylated Tau (p-Tau)) have a high sensitivity and specificity for AD, but
there is still no test to effectively predict the development of AD in a
pre-symptomatic stage [259,260]. Therefore, AD biomarkers are urgently needed
for both early and accurate diagnosis and prediction of disease progression.
Among peripheral candidate proteins, most AD-related proteins reported in the
literature are not specific and were found to be affected by other brain
disorders [261]. Aβ
is the main constituent of senile plaques in AD. Measurement of Aβ1-42 in CSF is a valuable marker
in AD research, where low levels indicate AD [259]. For the past 5 years some
new biomarkers have been postulated as potential diagnostic and/or prognostic
candidates for AD. A novel serum proteomic approach to interrogate the
low-molecular weight proteome for serum AD found 59 novel potential AD
biomarkers, 4 of which showed diagnostic replicability [262]. Panel-based
proteomics on plasma samples from Twins-UK subjects revealed that genetic
factors explain ~26% of the variability in blood protein levels on average. The
plasma level of the mitogen-activated protein kinase (MAPK) MAPKAPK5 protein
was found to positively associate with the 10-year change, and the plasma level
of protein MAP2K4 was found to suggestively associate negatively with the
volume of the left entorhinal cortex [263]. Multiple markers were identified to
be differentially expressed in the CSF of AD patients as compared with control subjects.
Two of these novel markers are neuronal secretory protein VGF and neuronal
pentraxin receptor-1 (NPTXR), which are decreased in AD (at baseline, 21% and
17%, respectively), with a decrased rate/year of 10.9% and 6.9%, respectively
[264].
Failures in the ubiquitin-proteasome system (UPS) during aging may
contribute to cellular stress and AD pathogenesis. Protein ubiquitination is
one of the key modulators of AD. Mutations in ubiquitin B mRNA that result in
UBB+1 dose-dependently cause an impaired UPS, subsequent
accumulation of UBB+1 and depositions of aberrant proteins in
plaques and tangles. Nuclei with substantial accumulations of tangle-bearing
neurons, such as the nucleus basalis of Meynert and raphe nuclei also present
high densities of UBB+1-positive tangles. Areas outside the
forebrain are also affected in AD [265].
Aβ interacts with
a variety of Aβ-associated
proteins (AAPs), some of which can form complexes with Aβ and influence its clearance,
aggregation or toxicity. The secreted Wnt pathway protein Dickkopf-related
protein 3 (Dkk-3) is a potential Aβ-associated protein. Dkk-3 co-localizes with Aβ in the brain, is expressed in
neurons and in blood vessel walls, is secreted by leptomeningeal smooth muscle
cells in vitro, and is abundantly present in both cerebrospinal fluid and
serum, but its levels are similar in non-demented controls and patients with
AD, Lewy body dementia, and frontotemporal dementia [266].
Patients with mild cognitive impairment (MCI) who are converted into AD
cases have an abnormal CSF glycosylation profile. CSF glycosylation changes may
occur before the onset of the disease. Glycosyltransferase GnT-III might be
involved in AD inducing specific sugar modifications in the BACE-1 glycoprotein
[267].
Using cell-type specific proteomics of microdissected temporal cortex
neurons from patients with AD, 400 proteins have been identified, of which 78%
were neuronal and 50% were associated with AD [268]. Changes in phosphorylation
levels were found in 19 proteins involved in energy metabolism, neuronal
plasticity, signal transduction, and oxidative stress response in the parietal
cortex of AD patients at different stages of the disease [269]. Histone
post-translational modifications (PTMs) have been found in the frontal cortex
of AD patients. Decreases in methylation of H2B residue K108 (25 %) and H4
residue R55 (35 %) were detected. A 91 % increase in ubiquitination
of K120 on H2B was observed as well as an apparent loss in acetylation of the
region near the N-terminus of H4. This study, reported by Anderson and Turko
[270] is the first to demonstrate changes in methylation of H2B K108,
methylation of H4 R55, and ubiquitination of H2B K120 in frontal cortex from
human donors with AD.
Transgenic AβPPswe/PS1dE9
mice express a chimeric mouse/human amyloid-β protein precursor (Mo/HuAβPP695swe) and mutant human
presenilin 1 (PS1-dE9) associated with early-onset AD. 15 proteins are
significantly different between the AβPPswe/PS1dE9 mice and age-matched controls. The expression levels of
the following proteins in AβPPswe/PS1dE9
mice were found to be at least 1.5 times higher than those in normal mice:
DCC-interacting protein 13-beta, serum albumin, creatine kinase B-type, heat
shock 70 kDa protein 1A, T-complex protein 1 subunit beta, adenylate kinase
isoenzyme 1, pyruvate dehydrogenase E1 component subunit beta mitochondrial,
and V-type proton ATPase catalytic subunit A. The expression levels of other
proteins (dihydropyrimidinase-related protein 2, actin cytoplasmic 2, isoform 1
of V-type proton ATPase catalytic subunit, tubulin alpha-1C chain,
F-actin-capping protein subunit alpha-2, ubiquitin carboxyl-terminal hydrolase
isozyme L1, and actin cytoplasmic 1) were lower in the transgenic model. These
proteins are involved in regulating various cellular functions, including
cytoskeletal structure, energy metabolism, synaptic components, and protein
degradation [271]. Using a redox-proteomic approach, twelve proteins were found
to be significantly altered in the levels of protein carbonyls in the
hippocampus. These proteins are crucial in energy metabolism, protein folding,
cell structure, signal transduction and excitotoxicity. Increased expression
level of carbonyl reductase 1 (CBR1) and protein carbonyls have been observed
in the hippocampi of 3×Tg-AD mice before the appearance of Aβ plaques and neurofibrillary
tangles (NFTs). By redox proteomics, twelve specifically carbonylated proteins
were identified. Among them, alpha-enolase (ENO1) and glutamine synthetase (GS)
were identified as the common targets of oxidation in the brains of 3×Tg-AD
mice, mild cognitive impairment (MCI) sufferers and AD patients. The oxidation
of t-complex protein 1 subunit epsilon (CCT5) and protein disulfide-isomerase
A3 (PDIA3) were reported to be associated with AD [272]. In a study of the
human brain-insoluble proteome in AD by mass spectrometry, 4,216 proteins have
been identified, among which 36 proteins accumulate in the disease, including
U1-70K and other U1 small nuclear ribonucleoprotein (U1 snRNP) spliceosome
components. Similar accumulations in mild cognitive impairment cases indicate
that spliceosome changes occur in early stages of AD. Multiple U1 snRNP
subunits form cytoplasmic tangle-like structures in AD but not in other
examined neurodegenerative disorders, including PD and frontotemporal lobar
degeneration. Comparison of RNA from AD and control brains reveals dysregulated
RNA processing with accumulation of unspliced RNA species in AD, including myc
box-dependent-interacting protein 1, clusterin, and presenilin-1. U1-70K
knockdown or antisense oligonucleotide inhibition of U1 snRNP increases the
protein level of amyloid precursor protein, indicating unique U1 snRNP
pathology and implication of abnormal RNA splicing in AD pathogenesis [273].
Brain tissue from diabetic patients with cerebrovascular dementia or AD
contains significant deposits of oligomerized amylin. Amylin is a pancreatic
hormone that has amyloidogenic and cytotoxic properties similar to the Aβ peptide. Amylin is overexpressed
in patients with pre-diabetic insulin resistance or obesity leading to amylin
oligomerization and deposition in pancreatic islets. Amylin oligomerization was
implicated in the apoptosis of the insulin-producing β-cells, and may contribute to
cognitive dysfunction [274].
The tau protein is central to the etiology of several tauopathies (AD,
frontotemporal dementia, progressive supranuclear palsy, post-traumatic
dementia). Tau protein associates with the ribonucleoproteome, including major
protein complexes involved in RNA processing and translation, and binds to
several heat shock proteins, the proteasome-and microtubule- associate
proteins. Expression of P301L mutant tau disrupts interactions of the
C-terminal half of tau with heat shock proteins and the proteasome. Higher
propensity of P301L mutant tau to aggregate may reflect a perturbation of its
chaperone-assisted stabilization and proteasome-dependent degradation [275].
MAP2c prevents arachidonic acid-induced in vitro aggregation of tau. MAP2c
possesses chaperone-like activity while tau does not. Phosphorylation impairs
the chaperone activity of MAP2c, implying a crucial role of chaperone in
preventing tau fibrillation. MAP2c/MAP2 might be one of the regulators
maintaining tau homeostasis in the cell [276]. A major step forward in
understanding the role of Tau truncation would be to identify the precise
cleavage sites of the several truncated Tau fragments that are present in AD
brains, especially those truncated at the N-terminus. The Gln124-Tau fragment
displays a stronger ability to bind and stabilize microtubules, suggesting that
the Tau N-terminal domain could play a direct role in the regulation of
microtubule stabilization [277].
Protein expression profiles of patients with vascular dementia (VaD)
distinguished, from a total of 144 differentially expressed proteins,
upregulated proteins enriched in 2 subpathways of 1 pathway, and downregulated
proteins enriched in 162 subpathways of 36 pathways [278].
At present, over 1,000 different proteins have been identified in
proteomic analysis of AD cases; however, interpretation of results is
difficult, and a direct connection between specific proteomic profiles and AD
pathogenesis is still undefined.
Parkinson’s disease
Parkinson's disease (PD) is the second most common age-related
neurodegenerative disorder in which genomic, environmental, cerebrovascular,
and epigenetic factors are involved [279]. PD is characterized by progressive
degeneration of dopaminergic neurons in the substantia nigra pars compacta and
presence of α-synuclein-containing
protein aggregates. Over 100 genes might be involved in PD genomics of which at
least 15 PD loci (PARK1-15) and other
loci (a-synuclein, leucine-rich repeat
kinase 2, parkin, PTEN-induced putative kinase 1, DJ-1,ATP13A2) might be
causative [280]. Genetic etiology of PD associated with mutations in the SNCA, PARK2, PINK1, PARK7 and LRRK2 genes, whereas other loci (e.g. LRRK2, MAPT, SCA1, SCA2, spatacsin, POLG1,
GBA) might be susceptibility genes associated with sporadic PD without
family history [160,281]. Some studies suggest that common and rare genetic
variation in the PARK10 locus do not
influence the risk or age at onset of clinical PD [282].
Retromer is a protein assembly that plays a central role in
orchestrating export of transmembrane-spanning cargo proteins from endosomes
into retrieval pathways destined for the Golgi apparatus and the plasma
membrane. A specific mutation in the retromer component VPS35, VPS35 (D620N)
might link retromer dysfunction to familial autosomal dominant and sporadic PD.
In cells expressing VPS35 (D620N) there is a perturbation in endosome-to-TGN
transport but not endosome-to-plasma membrane recycling. The major defect of
the D620N mutation lies in the association to the actin-nucleating Wiskott-Aldrich
syndrome and SCAR homolog (WASH) complex, andthe primary defect of the VPS35
(D620N) mutant is a decrease in affinity for the WASH complex component FAM21
[283].
Parkin is an E3-protein ubiquitin ligase, which plays an important role
as a scavenger in cell metabolism. PARK3
mutations are associated with PD. The proteomic analysis of the mutant form of
the Parkin protein (Q311R and A371T), isolated from a PD patient, exhibited
anomalies at the proteome level probably due to the differences in processing
[284].
Genome-wide association studies have demonstrated association between
SNCA variability and susceptibility to PD. Risk variants affect methylation of
a putative promoter in SNCA intron 1. PD patients show significant
hypomethylation as compared with controls in blood samples, and rs3756063 is associated with SNCA methylation level in both blood and
brain [285]. Methylation of the α-synuclein (SNCA) gene may be
involved in PD pathogenesis due to altered gene expression, protein structural
changes and overexpression, and protein aggregation [286]. Methylation of SNCA intron 1 is associated with
decreased SNCA transcription. SNCA
hypomethylation is observed in the substantia nigra of sporadic cases,
accompanied by an increased SNCA
expression [287]. Increased α-synuclein production might be the result of increased SNCA expression due to hypomethylation
of the SNCA gene [286]. Furthermore, α-synuclein sequesters DNMT1,
leading to DNA hypomethylation in PD and dementia with Lewy bodies [288], and
overexpression of DNMT1 restores nuclear DNMT1 [286].
Masliah et al. [289] identified 10 genes among the top 1000 members of
the aging-related methylation module which were associated with PD (SLC12A5,
ABCA3, FHIT, FAT1, CPLX2, APBA1, MAGI2, CNTNAP2,
ATP8A2, SMOC2). MRI1 and TMEM9 were candidate genes
with increased methylation, and the GSST1, TUBA3E and KCNH1
genes showed decreased methylation. Methylation of the HLA-DRB1, LRKK1, MMEL1, HLA-DQB1, OR12D3 and VAV2 genes exhibited confusing results. A methylation-based EWAS
in PD patients identified 20 unique genes with a sizable difference in
methylation between PD and controls, while 17 were identified between PD with
anxiety and PD without anxiety. FANCC
cg14115740 and TNKS2 cg11963436
showed significant differential methylation between PD cases and controls
[290].
In experimental models (1-methyl-4-phenylpyridinium (MPP+),
paraquat, rotenone), inducing overexpression of human α-synuclein, α-synuclein translocates into the
nucleus interacting with histones and inhibiting histone acetylation, and
nuclear-targeted α-synuclein
binds to histones and reduces histone 3 acetylation through its association
with HDAC1 and SIRT2 [28,291-292].
Oxidative stress is a potential pathogenic mechanism in sporadic PD.
c-Abl plays an important role in oxidative stress-induced neuronal cell death.
C-Abl, a nonreceptor tyrosine kinase, is activated in an
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP)-induced acute
PD model. Conditional knockout of c-Abl in neurons or treatment of mice with
STI571, a c-Abl family kinase inhibitor, reduced the loss of dopaminergic
neurons and ameliorated the locomotive defects induced by short-term MPTP
treatment. p38α is a major
substrate of c-Abl and c-Abl-mediated phosphorylation is critical for the
dimerization of p38α.
p38α inhibition
mitigates the MPTP-induced loss of dopaminergic neurons [293]. The oxidative
stress-sensitive protein kinase Cδ (PKCδ) has been
implicated in dopaminergic neuronal cell death. The PKCδ gene can be regulated by histone
acetylation. Treatment with histone deacetylase (HDAC) inhibitor sodium
butyrate (NaBu) induces PKCδ
expression in cultured neurons, brain slices, and animal models.
Hyperacetylation of histone H4 by NaBu is associated with the PKCδ promoter. Deletion analysis of
the PKCδ promoter mapped the
NaBu-responsive element to an 81-bp minimal promoter region. Four GC boxes
conferred hyperacetylation-induced PKCδ promoter activation. Sp1, Sp3, and Sp4 regulate NaBu-induced PKCδ up-regulation. NaBu does not
alter the DNA binding activities of Sp proteins or their expression.
Overexpression of the p300/cAMP-response element-binding protein-binding
protein (CBP) potentiates the NaBu-mediated transactivation potential of
Sp1/Sp3, but expressing several HDACs attenuated this effect, suggesting that
p300/CBP and HDACs act as coactivators or corepressors in histone
acetylation-induced PKCδ
up-regulation. NaBu up-regulation of PKCδ sensitizes neurons to cell death in a human dopaminergic cell model
and brain slice cultures. Histone acetylation regulates PKCδ expression to augment
nigrostriatal dopaminergic cell death, which could contribute to the
progressive pathogenesis of PD [294].
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene, but not in its closest paralog LRRK1, have been linked to autosomal dominant PD. Epidermal growth
factor receptor (EGF-R) is a LRRK1-specific
interactor, while 14-3-3 proteins are LRRK2-specific.
LRRK1 and LRRK2 can carry out distinct functions by interacting with
different cellular proteins [295]. The most prevalent mutation, G2019S, results
in enhanced LRRK2 kinase activity that potentially contributes to the etiology
of PD. Disease progression might be mediated by poorly characterized
phosphorylation-dependent LRRK2 substrate pathways. 776 phosphorylation sites
were found to be increased or decreased by at least 50% in response to LRRK2
kinase inhibition (LRRK2-IN-1) treatment, including sites on proteins
previously known to associate with LRRK2. LRRK2-IN-1 inhibited
lipopolysaccharide-induced tumor necrosis factor alpha (TNFα) and C-X-C motif chemokine 10
(CXCL10) levels in astrocytes and also enhanced multiple neurite
characteristics in primary neuronal cultures. LRRK2-IN-1 had almost identical
effects in primary glial and neuronal cultures from LRRK2 knockout mice. LRRK2-IN-1 may inhibit pathways of perceived
LRRK2 pathophysiological function independently of LRRK2 [296].
Classical activation (M1 phenotype) and alternative activation (M2
phenotype) are the two polars of microglial activation states that can produce
either detrimental or beneficial effects in the CNS. Histone H3K27me3
demethylase Jumonji domain containing 3 (Jmjd3) is essential for M2 microglia
polarization. Suppression of Jmjd3 in N9 microglia inhibits M2 polarization and
simultaneously magnifies M1 microglial inflammatory responses, which leads to
extensive neuron death in vitro. The suppression of Jmjd3 in the substantia
nigra (SN) causes microglial overactivation and exacerbates dopamine (DA)
neuron death in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine
(MPTP)-intoxicated mouse model of PD. Jmjd3 levels are lower in the midbrain of
aged mice, where H3K27me3 levels are increased, as well as the M1/M2 ratio,
suggesting that aging is a contributing factor in switching the microglia
phenotypes. In the PD context, Jmjd3 is able to enhance the polarization of M2
microglia by modifying histone H3K27me3, switching the microglia phenotypes
that might influence neuroimmune dysfunction in PD [297].
LncRNAs are also important players in the pathoepigenetics of PD [33].
Nuclear factor E2-related factor 2 (Nrf2) is a key transcription factor that
regulates the expression of antioxidant and detoxifying genes that provide
cellular protection against various stressors, including reactive oxygen
species (ROS). Nrf2 activity is tightly regulated by a cytoplasmic inhibitory
protein Kelch-like ECH-associated protein 1 (Keap1). microRNA-7 (miR-7), which
is highly expressed in the brain, represses Keap1 expression by targeting the
3'-untranslated region (UTR) of its mRNA in human neuroblastoma cells, SH-SY5Y.
Targeted repression of Keap1 and activation of Nrf2 pathway underlies the
cytoprotective effects of miR-7 against 1-methyl-4-phenylpyridinium (MPP+)-induced
toxicity in SH-SY5Y and differentiated human neural progenitor cells, ReNcell
VM [298].
Some neuronal processes involve the mechanistic/mammalian target of
rapamycin complex 1 (mTORC1). Activation of mTORC1 promotes translation.
Curtailing the activity of mTORC1 bidirectionally alters the expression of
proteins associated with epilepsy, AD, ASD, and PD.The protein expression of
PARK7 is sensitive to mTORC1 inhibition. In a mouse model of tuberous sclerosis
complex (TSC), with overactive mTORC1 signaling, PARK7 protein is elevated in
the dendrites and colocalizes with the postsynaptic marker PSD-95 [299].
Quantitative proteomics of protein expression profiles in the nigral tissue of
PD patients and control subjects revealed the presence of 11 differentially
expressed proteins, including alphaB-crystallin (Cryab). Cryab was markedly
upregulated in the SN of PD brain. Cryab expression was also upregulated in
reactive astrocytes and microglia in a neurotoxin-induced mouse PD model.
Increased expression of Cryab was also present in cytoplasmic inclusions in a
subset of glial cells in Parkinsonian brains, suggesting that Cryab may be
involved in the glial pathology during dopaminergic neuron degeneration in PD
[300].
Oxidative stress and mitochondrial dysfunction may be involved in the
pathogenesis of PD. Protein alterations
in PD brain are dominated by mitochondrial and lipid transport defects, and are
largely independent of transcriptional changes [301]. Mutations in the
mitochondrial Ser/Thr kinase PTEN-induced kinase 1 (PINK1) are associated with an autosomal recessive familial form of
early-onset PD. PINK1 plays important neuroprotective roles against
mitochondrial dysfunction by phosphorylating and recruiting Parkin, a cytosolic
E3 ubiquitin ligase, to facilitate elimination of damaged mitochondria via
autophagy-lysosomal pathways. Loss of PINK1 in cells and animals leads to
various mitochondrial impairments and oxidative stress, culminating in
dopaminergic neuronal death in humans. Changes in the brain proteome and
phosphoproteome of mice lacking PINK1
suggest that defects in signaling networks, energy metabolism, cellular
proteostasis, and neuronal structure and plasticity are involved in the
pathogenesis of familial PD. Changes in the proteome and phosphoproteome of the
PINK1 knockout mouse brain revealed
alterations in key proteins associated with increased oxidative stress,
aberrant cellular signaling, altered neuronal structure, decreased synaptic
plasticity, reduced neurotransmission, diminished proteostasis networks, and
altered metabolism, including 14-3-3ε (14-3-3 protein epsilon), 3-PGDH (phosphoglycerate dehydrogenase),
ALDOA (aldolase A), APT1 (acyl-protein thioesterase 1), CaM (calmodulin), CBR3
(carbonyl reductase [NADPH] 3), ENO2 (gamma-enolase), HPRT(hypoxanthine-guanine
phosphoribosyltransferase), HSP70(heat-shock-related 70 kDa protein 2),
IDHc (cytoplasmic isocitrate dehydrogenase [NADP+]), MAPK1 (mitogen-activated
protein kinase 1), MEK1 (MAP kinase kinase 1), MDHc (cytoplasmic malate
dehydrogenase), NFM (neurofilament medium polypeptide), NSF
(N-ethylmaleimide-sensitive fusion protein), PHB (prohibitin), PINK1
(PTEN-induced putative kinase 1), PPIaseA (peptidyl-prolyl cis-trans isomerase
A), PSA2 (proteasome subunit alpha type-2), TK (transketolase), and VDAC-2
(voltage-dependent anion-selective channel protein 2) [302]. The proteomic
analysis of the PINK1 kinase-PARKIN UB ligase mitochondrial control pathway
disrupted in PD suggests that PINK1 plays a dual role by phosphorylating PARKIN
on its UB-like domain and poly-UB chains on mitochondria. PARKIN activation by
PINK1 produces canonical and noncanonical UB chains on mitochondria, and
PARKIN-dependent chain assembly is required for accumulation of poly-phospho-UB
(poly-p-UB) on mitochondria [303]. Mitochondrial complex I impairment in PD is
modeled in vitro by the susceptibility of dopaminergic neurons to the complex I
inhibitor 1-methyl-4-phenylpyridinium (MPP+). microRNA-7 (miR-7),
which is expressed in tyrosine hydroxylase-positive nigral neurons in mice and
humans, protects cells from MPP+-induced toxicity in dopaminergic
SH-SY5Y cells, differentiated human neural progenitor ReN cell VM cells, and
primary mouse neurons. RelA, a component of nuclear factor-κB (NF-κB), was identified to be
downregulated by miR-7 using quantitative proteomic analysis. RelA mRNA is a
target of miR-7 and is required for cell death following MPP+
exposure. RelA mediates MPP+-induced suppression of NF-κB activity, which is essential for
MPP+-induced cell death. The protective effect of miR-7 is exerted
through relieving NF-κB
suppression by reducing RelA expression [304]. Mitochondrial transcription
factor A (TFAM) regulates mitochondrial biogenesis, which is downregulated by
extracellular signal-regulated protein kinases (ERK1/2) in cells treated
chronically with the complex I inhibitor MPP+. Mutation of TFAM at
serine 177 to mimic phosphorylation recapitulated the effects of MPP+
in decreasing the binding of TFAM to the light strand promoter, suppressing
mitochondrial transcription. Mutant TFAM was unable to affect respiratory
function or rescue the effects of MPP+ on respiratory complexes
[305]. Mitochondrial protein profiles during dopaminergic neuronal cell death
(MN9D) induced by 6-hydroxydopamine (6-OHDA) revealed several protein
candidates among which TNF receptor-associated protein 1 (TRAP1), a
mitochondrial molecular chaperone, was released from the mitochondria into the
cytosol in MN9D cells [306].
The proteomic analysis of chronic
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced non-human primate
animal model of parkinsonism allowed the identification of 86 glycoproteins,
163 non-glycoproteins, and 71 phosphoproteins differentially expressed in the
MPTP-treated monkeys [307]. Microarray and proteomic data have revealed
abnormal expression of several genes and proteins responsible for PD.
Unreported 37 PD disease markers were identified based on their topological
significance in the networks. Of these 37 markers, 8 were significantly
involved in the core functional modules and showed significant change in
co-expression levels, and 4 (ARRB2, STX1A, TFRC and MARCKS) were found to be
associated with several neurotransmitters, including dopamine [308].
Several candidate biomarkers in biological fluids are being pursued as
blood-based biomarkers in PD (α-synuclein, DJ-1, uric acid, epidermal growth factor,
apolipoprotein-A1, and peripheral inflammatory markers) [309]. Proteomic
profiling followed by high-throughput targeted mass spectrometry (MS), in order
to identify peptides in human cerebrospinal fluid (CSF) for PD diagnosis and
disease severity correlation, characterized 14,000 unique peptides displaying
differences between PD and healthy controls. Specifically, 5 peptides derived
from SPP1, LRP1, CSF1R, EPHA4, and TIMP1 were identified with a sensitivity of
76.7% and a specificity of 80.0% for PD. A combination of two peptides
belonging to proteins TIMP1 and APLP1 significantly correlated with disease
severity [310]. Using a high-throughput shotgun proteomic strategy, 1795 nigral
proteins were identified. Of them, 204 proteins displayed significant
expression level changes in PD patients versus controls. These were involved in
novel or known pathogenic processes including mitochondrial dysfunction,
oxidative stress, or cytoskeleton impairment. The differential expression of
ferritin-L and seipin was confirmed, and the neuronal localization of gamma
glutamyl hydrolase and nebulette was demonstrated, suggesting a role for
nebulette overexpression in PD neurodegeneration [311].
Trinucleotide repeat
disorders
Polyglutamine disorders (Table 1)
are caused by expansion of CAG trinucleotide repeats encoding polyglutamine
tracts in specific genes [312]. The family of polyglutamine diseases includes
spinal and bulbar muscular atrophy (SBMA), Huntington’s disease (HD),
dentatorubral-pallidoluysian atrophy (DRPLA), and spinocerebellar ataxia (SCA)
type 1, 2, 3, 6, 7, and 17. These disorders are caused by glutamine expansions
in androgen receptor (AR), huntingtin, atrophin-1, ataxin-1, ataxin-2,
ataxin-3, CACNA1A, ataxin-7, and the TATA-box binding protein (TBP),
respectively [312,313]. Polyglutamine expansion in the androgen receptor (AR)
is responsible for spinobulbar muscular atrophy (SBMA) that leads to selective
loss of lower motor neurons. Protein arginine methyltransferase 6 (PRMT6) is a
specific co-activator of normal and mutant AR and the interaction of PRMT6 with
AR is significantly enhanced in the AR mutant. AR and PRMT6 interaction occurs
through the PRMT6 steroid receptor interaction motif, LXXLL, and the AR
activating function 2 surface. AR transactivation requires PRMT6 catalytic
activity and involves methylation of arginine residues at Akt consensus site
motifs, which is mutually exclusive with serine phosphorylation by Akt. The
enhanced interaction of PRMT6 and mutant AR leads to neurodegeneration in cell
and fly models of SBMA, indicating a direct role of arginine methylation in
polyglutamine disease pathogenesis [313].
A prototypal example of neurodegeneration, in which genomic and
epigenomic alterations coexist, is Huntington’s chorea-related striatal
degeneration, characterized by: (i) mutations (CAG expansions) in the
huntingtin (HTT) gene; (ii) mutant
HTT-related excitotoxicity, mitochondrial dysfunction, axonal transport
deficit, altered proteasome activity, and gene dysregulation; (iii)
dysregulation of multiple genes; (iv) interference of nuclear localization of
expanded HTT with transcription factors, co-activators, and proteins of the
transcriptional machinery; (v) alteration of cytoplasmic retention of the
transcriptional repressor REST, which is normally associated with wild-type
HTT; (vi) alteration of transcription of multiple genes involved in neuronal
survival, plasticity, signaling, and mitochondrial biogenesis and respiration;
(vii) dysmorphic chromatin structure through altered post-translational
modifications of histones and methylation of DNA; (viii) multiple alterations
of histone post-translational modifications, including acetylation,
methylation, ubiquitylation, polyamination, and phosphorylation; (ix) altered
expression and regulation of non-coding miRNAs controlled by REST; and (x)
concomitant de-repression of downstream mRNA targets [314,315].
Pathogenic CAG (cytosine-adenine-guanine) expansions beyond certain
thresholds in the ataxin-2 (ATXN2)
gene cause spinocerebellar ataxia type 2 (SCA2)
and also contribute to PD, amyotrophic lateral sclerosis, and frontotemporal
lobar degeneration. ATXN2 levels are
controlled by DNA methylation which influences age at onset and anticipation
[316].
Other neurological
disorders
Epigenetic and proteomic changes are frequently seen in many other CNS
disorders. Transmissible encephalopathies (TSEs), such as Creutzfeldt-Jakob
disease (CJD) and scrapie, are caused by prions that provoke strain-specific
patterns of disease. Misfolded host prion protein (PrP-res amyloid) is believed
to be the causal infectious agent. Kipkorir et al. [317] have identified host
proteins bound with FU-CJD agent infectious brain particles by proteomic
analysis. More than 98 proteins were differentially regulated, and 56 FU-CJD
exclusive proteins were revealed after PrP, GFAP, C1q, ApoE, and other late
pathologic response proteins were removed. Stripped FU-CJD particles revealed
HSC70, cyclophilin B, an FU-CJD exclusive protein required by many viruses, and
early endosome-membrane pathways known to facilitate viral processing,
replication, and spread. Synaptosomal elements including synapsin-2 and AP180
paralleled the known ultrastructural location of 25 nm virus-like TSE particles
and infectivity in synapses. Human sCJD brain particles contain 146 exclusive
proteins, in addition to heat shock, synaptic, and viral pathways, and
Alzheimer, Parkinson, and Huntington aggregation proteins.
Frontotemporal lobar degeneration (FTLD) comprises a spectrum of
uncommon neurodegenerative diseases with an estimated prevalence of 15-22 cases
per 100,000 persons. This cluster of related dementias include the behavioral
variant of frontotemporal dementia (bvFTD), progressive non-fluent aphasia
(PNFA), semantic dementia (SD), FTD with motor neuron disease (FTD-MND),
progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS).
Intracellular protein aggregates are the major neuropathological hallmark of
FTLD, suggesting the presence of abnormal protein metabolism or function in the
disease's pathogenesis. Studies of CSF proteomics provided a set of candidate
biomarkers which need further validation [318].
DNA methylation plays a key role in cell fate determination. DNA
methylation at the 5 position of cytosine (5-mC) is an epigenetic marker of
biological and pathological processes. 5-mC can be converted to
5-hydroxymethylcytosine (5-hmC) by the ten-eleven translocation (TET) family
proteins, which is now recognized as the "sixth base" in the
mammalian genome, following 5-mC, the "fifth base". 5-hmC is present
in brain, embryonic stem cells, and many other tissues. 5-hmC and the TET
family proteins might be involved in gene control mechanisms, DNA methylation
regulation, and in the pathophysiology of human disease [319]. Cell-specific
increases of 5mC and 5hmC are associated with the death of retinal neurons
during both development and degeneration, suggesting that changes in DNA
methylation may play a role in modulating gene expression during the process of
retinal degeneration. During retinal development, hypermethylation of retinal
neurons associates with classical caspase-dependent apoptosis as well as
caspase-3 independent cell death, while hypermethylation in the rd1 mouse
photoreceptors is primarily associated with caspase-3-independent programmed
cell death [320].
Loss of 5hmC is a hallmark of human malignancies (glioma, melanoma,
myeloid tumors). In myeloid malignancies, loss of 5hmC is due to mutations
within ten-eleven-translocation (TET)
genes, enzymes being responsible for conversion of 5mC to 5hmC. There are TET2 and TET3 alterations in human gliomas. Kraus et al. [321] identified 7
genetic alterations within TET2 (p.V218M, p.G355N, p.P363L, p.L1721W, p.P1723S,
p.I1762V, p.H1778R). In contrast to leukemia, loss of 5hmC in glioma is not
caused by TET gene alterations.
Disrupted gene expressions or functional inhibitions of TET proteins might be
responsible for the aberrant epigenome of human glioma. Subtelomeric regions
dynamically change their epigenetic pattern during development and progression
of several malignancies and degenerative disorders. DNA methylation levels
dramatically increase at the subtelomere of Chr.8q, 21q, and XpYp in malignant
glioma [322].
The Polycomb group (PcG) proteins play a critical role in
histone-mediated epigenetics which has been implicated in the malignant
evolution of glioblastoma multiforme (GBM). Li et al. [323] found widespread
aberrant expression of the PcG members in GBM samples compared to normal brain,
including upregulation of EZH2, PHF19,
CBX8 and PHC2 and downregulation
of CBX7, CBX6, EZH1 and RYBP. Changes in EZH2, PHF19, CBX7, CBX6 and
EZH1 occurred progressively as astrocytoma grade increased.
Brain metastatic disease is a common phenomenon in patients with breast
cancer. Genomic and epigenomic events underlie breast cancer brain metastasis.
Large chromosomal gains in 1q, 5p, 8q, 11q, and 20q, broad-level deletions
involving 8p, 17p, 21p and Xq, amplified and overexpressed genes (ATAD2, BRAF, DERL1, DNMTRB and NEK2A), and deleted or underexpressed
genes (ATM, CRYAB, HSPB2) are
frequently seen in this clinical condition. Enrichment in cell cycle and G2/M
transition pathways, which contained AURKA,
AURKB, and FOXM1, are also
frequent. While overall methylation levels are increased in breast cancer brain
metastasis, basal-like brain metastases are associated with lower levels of
methylation. Integrating DNA methylation data with gene expression revealed
defects in cell migration and adhesion due to hypermethylation and
downregulation of PENK, EDN3, and ITGAM. Hypomethylation and upregulation
of KRT8 probably affects adhesion and
permeability, as reported by Salhia et al. [324].
Nguyen [325] identified epigenetic regulation of alternative APP pre-mRNA splicing in the Lesch-Nyhan
syndrome (LNS), showing for the first time the presence of several APP-mRNA isoforms encoding APP protein
isoforms ranging from 120 to 770 amino acids (with or without mutations and/or
deletions). New APP-mRNA isoforms
with a deletion followed by an insertion (INDELS) in LNS and LNVs patients have
been identified, suggesting a role for genomic rearrangements of APP gene via the Fork Stalling and
Template Switching (FoSTeS) mechanism leading to INDELS. Epistasis between
mutated HPRT1 and APP genes could be one of the factors of
epigenetic modifications responsible for genomic rearrangements of the APP gene.
The type III histone deacetylase sirtuin 1 (Sirt1) has recently emerged as a critical immune regulator by
suppressing T cell immunity and macrophage activation during inflammation. Mice
with genetic Sirt1-deletion
specifically in dendritic cells (DCs) are resistant to MOG-induced experimental
autoimmune encephalomyelitis (EAE). Loss of Sirt1 functions in DCs enhances
their ability to produce IL-27 and interferon (IFN-β). Co-cultivation of
Sirt1-null DCs with CD4+ T cells inhibits Th17 differentiation,
which is reversed by anti-IL27 and anti-IFN-β neutralization antibodies. Sirt1
antagonizes acetylation of IRF1, a transcription factor that drives IL-27
production. Genetic deletion of IRF1
in Sirt1-null DCs abolishes IL-27
production and suppresses Th17 differentiation. These data indicate that the
histone deacetylase Sirt1 programs DCs to regulate Th17 differentiation during
inflammation [326].
Whole brain radiotherapy (WBRT) produces unwanted sequelae. Sirtuin 2
(SIRT2) is a deacetylase expressed in the CNS. It has been postulated that a
single disease pathway is responsible for radiation-induced brain injury in Sirt2 wild-type (WT) and knockout (KO)
mice at the proteomic level. Canonical pathways for Huntington's, Parkinson's,
and Alzheimer's disease were acutely affected by radiation within 72 h of
treatment. Although loss of Sirt2 preferentially affected both Huntington's and
Parkinson's pathways, WBRT most significantly affected Huntington's-related
proteins in the absence of Sirt2. Identical protein expression patterns were
identified in Mog following WBRT in both Sirt2 WT and KO mice, revealing a
proteomic radiation signature; however, long-term radiation effects were found
to be associated with altered levels of a small number of key
neurodegeneration-related proteins, identified as Mapt, Mog, Snap25, and Dnm1.
The presence of Sirt2 can have significant effects on the brain proteome and
its response to ionizing radiation [327].
Epigenetic changes also occur after nerve tissue injury. Epigenetic
regulation of CC-chemokine ligand (CCL)
2 and CCL3 participates in the
peripheral sensitization leading to neuropathic pain. Kiguchi et al. [328]
examined the relationship between histone H3 modification and the upregulation
of these molecules using a mouse model of neuropathic pain after partial
sciatic nerve (SCN) ligation (PSL). The mRNA levels of CCL2, CCL3 and their receptors (CCR2
and CCR1/CCR5, respectively) were
increased in the injured SCN. The levels of lysine 9-acetylated histone H3
(H3K9Ac) and lysine 4-trimethylated H3 (H3K4me3) in the promoter regions of the
CCL2 and CCL3 genes were increased in the injured SCN after PSL, indicating
the enhancement of gene expression. Upregulation of CCLs and CCRs was
suppressed by the histone acetyltransferase inhibitor, anacardic acid. These
chemokine cascades may subsequently elicit chronic neuroinflammation following
nerve injury.
Other neurological diseases in which epigenetic aberrations may be
involved include multiple sclerosis [329-331], migraine [332], epilepsy [333],
facioscapulohumeral muscular dystrophy [334-336], Duchenne muscular dystrophy
[337], chronic pain [338,339], Hutchinson-Gilfordprogeria syndrome [340-342],
genotoxic disorders [343,344], brain tumors [345], and stroke [346,347].
Epigenetic Mendelian
Disorders
Epigenetic
Mendelian disorders (EMD) are a group of multiple congenital anomaly and
intellectual disability syndromes resulting from mutations in genes encoding
components of the epigenetic machinery [5] (Table 2). Within this category, genetic mutations may affect
writers, erasers, or readers of epigenetic marks, and chromatin remodelers, as
well. Many EMD fall within the category of neurodevelopmental and imprinting
disorders, and some of them may manifest in adults. EMD involving the DNA
methylation machinery have been described for writers and readers of DNA
methylation: (i) Rett syndrome, an X-linked disorder affecting mostly females
and resulting from loss-of-function mutations in a reader of CpG methylation (MeCP2) (methy-CpG-binding protein); (ii)
2q23.1 microdeletion/microduplication syndrome, an autosomal dominant syndrome
with deletion/duplication in the MBD5
locus, encoding a methyl-CpG-binding protein; (iii) immunodeficiency,
centromeric instability, and facial anomalies (ICF) syndrome, caused by
homozygous or compound heterozygous hypomorphic mutations in the DNMT3B gene; (iv) hereditary sensory and
autonomic neuropathy with dementia and hearing loss (HSAN1E)(mutations in DNMT1 exon 20); (v) autosomal dominant
cerebellar ataxia, deafness, and narcolepsy (ADCADN)(mutations in DNMT1 exon 21). EMD of the histone
machinery have been described for writers, erasers, readers, and chromating
remodelers, including: (i) Kabuki syndrome, an autosomal dominant trait with
mutations in mixed lineage leukemia 2 (MLL2)(a
histone H3K4 methyltransferase) or lysine-specific demethylase 6A (KDM6A) genes; (ii) Rubinstein-Taybi
syndrome (RTS), an autosomal dominant syndrome caused by haploinsufficiency of
histone acetyltransferase enzyme genes (CREBBP
and EP300); (iii) Genitopatellar
syndrome (GPS) and Say-Barber-Biesecker-Young-Simpson (SBBYS) syndrome
(mutations in the histone acetyltransferase KAT6B),
(iv) Widerman-Steiner syndrome (WSS)(mutations in the MLL gene, histone methyltransferase H3K4), (v) Kleefstra syndrome
(KLFS)(mutations in EHMT1, histone
methyltransferase H3K9), (vi) Weaver syndrome (WS)(mutations in EZH2, histone methyltransferase H3K27),
(vii) Sotos syndrome (SS)(mutations in NSD1,
histone methyltransferase H3K36 and H4K20); (viii) brachydactyly-mental
retardation (BDMR) syndrome (haploinsufficiency of the histone deacetylase
gene, HDAC4); (ix) Cornelia de Lange
syndrome 5 (CDLS5) (X-lined) and Wilson-Turner syndrome (WTS)(X-linked)
(mutations in histone deacetylase HDAC8),
(x) Claes-Jensen syndrome (CJS) (X-lined)(mutations in KDMSC, histone demethylase H3K4); (xi) Kabuki syndrome
(X-linked)(mutations in KDM6A,
histone demethylase H3K27); (xii) Siderius X-linked mental retardation
sysndrome (MRXSSD) (mutations in PHF8,
plant homeodomain finger protein); (xiii) Börjeson-Forssman-Lehmann syndrome
(BFLS)(X-linked recessive trait, missense mutations in the PHF6 gene, plant homeodomain finger protein); and (xiv) X-linked
mental retardation and macrocephaly (mutations in BRWD3, bromodomain-containing protein). EMD of chromatin remodelers
include the following: (i) alpha-thalassemia/mental retardation X-linked
(ATRAX) syndrome (mutations in ATRAX, SWI/SNF
ATP-dependent chromatin remodeler); (ii) 4 variants of Coffin-Siris syndrome:
mental retardation autosomal dominan 14 (MRD14) (mutations in ARID1A), mental retardation autosomal
dominant 12 (MRD12) (mutations in ARID1B),
mental retardation autosomal dominant 16 (MRD16) (mutations in SMARCA4), and mental retardation
autosomal dominant 15 (MRD15) (mutations in SMARCB1);
(iii) Rhabdoid tumor predisposition syndrome 2 (mutations in SMARCA4); (iv) Schwannomatosis
(mutations in SMARCB1); (v) Rhabdoid
tumor predisposition syndrome 1 (mutations in SMARCB1); (vi) Nicolaides-Baraitser syndrome (mutations in SMARCA2); (vii) Floating harbor syndrome
(mutations in SRCAP, INO80/SWR1
ATP-dependent chromatin remodeler); (viii) CHARGE syndrome (mutations in CHD7, CHD ATP-dependent chromatin
remodeler); and (ix) mental retardation autosomal dominant 21 (MRC21)
(mutations in CTCF,
chromatin-organizing zinc finger protein) [5] (Table 2).
Future Trends
Most CNS disorders are clinical entities which, in many instances,
share some common features: (i) pathogenically, they are complex disorders in
which a plethora of plural events (genomic defects, epigenetic aberrations,
mitochondrial dysfunction, environmental factors) is potentially involved; (ii)
many of them, especially those with a late onset, are characterized by
intracellular and/or extracellular deposits of abnormal proteins; (iii) their
diagnosis is difficult because they lack specific biomarkers (and their
prediction is almost impossible); (iv) their treatment is symptomatic (not
anti-pathogenic) and not cost-effective; and (v) the vast majority represent
chronic ailments with progressive deterioration and bad prognosis [14]. The
concept of epigenetics, introduced by Conrad Waddington in 1942, and its
spectacular evolution, from a biotechnological perspective, has been of great
help for the past 10 years in the
understanding of gene regulation and expression (functional genomics),
neurogenomics, and pathogenetics of CNS disorders [2,17,28,166,177] (Figure 1).
Gene expression and protein function experience profound modifications
throughout the life span. It is likely that the frontier between health and
disease is not only associated with specific SNP variability and epigenetic
aberrations (in conjunction with environmental risks) but also with a
salutary/pathogenic threshold of transformed protein accumulation in critical
cells (especially in neurons). Over the past decade, progress in epigenetics
and proteomics has helped to understand many aspects of pathogenic phenomena
which remained obscure or unaffordable to our technical capabilities for the
assessment of genomic dysfunction, epigenetic dysregulation, and abnormal
protein expression. Transcription errors represent a molecular mechanism by
which cells can acquire disease phenotypes. The error rate of transcription
increases with cell aging, suggesting that transcription errors affect
proteostasis particularly in aging cells. Accordingly, transcription errors
accelerate the aggregation of peptides and shorten the lifespan of cells [348].
Novel methodologies have allowed us to
configure new pathogenic hypotheses for a better understanding of brain
disorders. In this endeavor, epigenetics and proteomics have been of great
benefit. Epigenetic studies have revealed the important role that epigenetic
modifications have on brain development and maturation, synaptic plasticity
[13], brain sex differences [43], neurodevelopment and imprinting disorders,
mental disorders [38], neurodegeneration [10,17], and the new field of
epigenetic Mendelian disorders [5]. Structural genomic defects cannot explain
in full the pathogenesis of CNS disorders. Many old concepts
related to the pathogenesis of CNS disorders should be eliminated. Parkinson’s
disease is not the result of a single deficiency in dopamine; Alzheimer’s
disease is not the consequence of a cholinergic deficit; however, the basic
principles for the development of the most currently prescribed drugs for both
disorders rely on a single neurotransmitter defect (enhancement of dopamine
neurotransmission in Parkinson’s disease, and potentiation of cholinergic
transmission with acetylcholinesterase inhibitors in Alzheimer’s disease).
These old-fashioned pathogenic concepts are completely out-of-date, and the new
conceptions on neurodegenerative disorders are based on the pathogenic cascade
represented by genomic-epigenomic-transcriptomic-proteomic-metabolomic
disturbances leading to a specific phenotype which in the future will require a
personalized therapeutic intervention (pharmacogenomics, pharmacoepigenomics)
for phenotype disease modification (Figure
1). The same must happen with most mental disorders whose
psychopharmacological treatments rely on a reductionistic view based on the
regulation of 6 neurotransmitters (dopamine, noradrenaline, serotonin, acetylcholine,
histamine, GABA), assuming for the past 50 years that major psychiatric
disorders (schizophrenia, depression, anxiety, bipolar disorder, autism,
attention deficit hyperactivity disorder) are merely neurotransmitter disorders
[69,83,84,349]. However, as pointed out by Riley et al. [301],
systems analysis is believed to help deconvolute complex biological responses
involving hundreds or thousands of genes assayed by OMICs methods. Although
systems-style approaches have been applied to CNS tissues, most studies have
used simple functional overview approaches resulting in the identification of
differentially expressed genes, or pathways. While these approaches expanded
our understanding of disease-related changes, they are not able to elucidate the
complex interconnectivity of biological and pathological processes present
within diseased tissue. These approaches are “low resolution” descriptive
methods with limited projection in terms of clarifying molecular pathogenesis,
experimental follow-up, and clinical application.
Global protein profiling by mass spectrometry-based proteomics has
evolved as a new hypothesis-free avenue to optimally unravel new candidate
protein biomarkers involved in different CNS disorders.Technological
developments and improvement of sensitivity, specificity and speed of different
proteomic approaches have facilitated the discovery of an enormous number of
biomarker candidates; however, most biomarkers have not yet been validated,
which limit their application in clinical practice. The correct interpretation
of thousands of data derived from proteomic and epigenomic analysis is an
additional problem for the practical implementation of biomarkers in the
clinical setting [350]. Novel neuroproteomic tools and powerful bioinformatic
resources are needed to accelerate the incorporation of proteomic and
epigenomic analysis to the diagnostic process [351-353].
Another important field, in which epigenetics and proteomics are
contributing to its expansion, is drug development. Epigenetic drugs are
becoming a fashion [9,10,354] and some of them have been approved by the FDA in
recent years for the treatment of cancer [19]. However, most epigenetic drugs
are pleiotropic and are not devoid of toxicity and biodynamic complications
(e.g. brain penetration) [9].
The effects of drugs (pharmacokinetics and pharmacodynamics) and their
therapeutic outcome in the treatment of a given disease are the result of a
network of metabolomic events (genomics-epigenomics-transcriptomics-proteomics)
associated with the binomial interaction of a chemical or biological molecule
with a living organism. The clusters of genes currently involved in a
pharmacogenomic process include pathogenic, mechanistic, metabolic,
transporter, and pleiotropic genes [14]. In practice, the expression of these
genes is potentially modifiable (transcriptionally and/or
post-transcriptionally) by epigenetic mechanisms which may alter (i) pathogenic
events, (ii) receptor-drug interactions, (iii) drug metabolism (phase I and II
enzymatic reactions), (iv) drug transport (influx-efflux across membranes and
cellular barriers), and (v) pleiotropic events leading to unexpected
therapeutic outcomes. The understanding of these mechanisms is the main focus
of pharmacoepigenomics in order to optimize therapeutics and advance towards a
personalized medicine [9,355].
In the coming years, important achievements must be accomplished in
different areas of neuroscience: (i) brain development and maturation, (ii)
toxicogenomics, (iii) functional epigenomics, (iv) proteoepigenomics, (v)
pathoepigenomics, (vi) predictive proteomics, (vii) diagnostic proteomics,
(viii) prognostic proteomics, (ix) pharmacoepigenomics, and (x)
epitherapeutics. It is likely that systems biology will dominate the –omics
signatures [356]. Relevant information obtained from the ENCODE Project will be
incorporated into a more versatile map of clinical neuroscience and practical
medicine [29,30,357]. Development is a dynamic process that involves interplay
between genes and the environment. Postnatal environment is shaped by
parent-offspring interactions that promote growth and survival and can lead to
divergent developmental trajectories with implications for later-life
neurobiological and behavioral characteristics [358]. The impact that
nutrition, emotions, drugs and environmental toxicants during prenatal
development may have on brain maturation and late CNS disorders requires urgent
clarification [359-361]. Important advances related to the role of epigenetics
in the pathogenesis of brain disorders will occur in the near future with reliable
applications. Predictive, diagnostic, and prognostic proteomics, as well as the
use of biomarkers to monitor the effects of drugs will experience a profound
change from the present inmature stage of the field to a more specific and
validated area with various applications in CNS disorders.
In therapeutics, important breakthroughs will occur in some of the
following areas: (i) epigenetic drug discovery for different CNS disorders and
cancer [9,10,27,193,362,363]; (ii) practical applications of pharmacogenomics
[14,364] and pharmacoepigenomics [365-368] for the optimization and
personalization of current drugs and new pharmacological treatments; (iii)
novel therapeutic approaches to decode and resolve potential resistance
mechanisms in cancer and psychiatric disorders [365,369,370]; and (iv)
targeting miRNAs in prevention and treatment of brain disorders [371-373].
CONCLUSIONS
- Epigenomic regulation is a
common phenomenon of gene expression control during development,
maturation and aging in physiological and pathological conditions.
- Classical epigenetic
mechanisms (DNA methylation, chromatin remodeling/histone modifications,
and miRNA regulation), are among the major regulatory elements that
control metabolic pathways.
- Preconceptional parental
exposure to environmental stimuli may determine the offspring’s phenotype
via heritable epigenetic mechanisms, and exposure to diverse external
elements may condition several categories of human diseases and CNS
disorders.
- Mutations in the genes
encoding elements of the epigenetic machinery can lead to epigenetic
Mendelian disorders.
- Epigenomic dysregulation
contributes to the pathogenesis of neurodevelopmental, imprinting, mental,
neurological, and neurodegenerative disorders.
- Some epigenetic aberrations
are conceptually reversible and can potentially be targeted by
pharmacological and dietary interventions.
- Proteomic biomarkers can be
usefull for both early and accurate diagnosis and prediction of CNS
disease progression.
- The correct interpretation of
thousands of data derived from proteomic and epigenomic analysis is still
a problem for the practical implementation of biomarkers in the clinical
setting. Novel neuroproteomic tools and powerful bioinformatic resources
are needed to accelerate the incorporation of proteomic and epigenomic
analysis to the diagnostic process.
- Epigenetic changes in genes
involved in pharmacogenomics (pathogenic, mechanistic, metabolic,
transporter, and pleiotropic genes) can also influence drug efficacy and
safety and drug resistance in brain disorders and cancer.
Proteomic
biomarkers, novel therapeutic approaches to decode and resolve potential drug
resistance mechanisms, and targeting miRNAs in prevention and treatment of
brain disorders are promising developments in the field of proteoepigenomics.
- Tammen SA, Friso S, Choi SW
(2013) Epigenetics: the link between nature and nurture. Mol Aspects Med
34: 753-764.
- Sweatt JD (2013) The emerging
field of neuroepigenetics. Neuron 80: 624-632.
- Harris RA, Nagy-Szakal D,
Kellermayer R (2013) Human metastable epiallele candidates link to common
disorders. Epigenetics 8: 157-163.
- Szulwach KE, Jin P (2014)
Integrating DNA methylation dynamics into a framework for understanding
epigenetic codes. Bioessays 36: 107-117.
- Fahrner JA, Bjornsson HT (2014)
Mendelian disorders of the epigenetic machinery: tipping the balance of
chromatin states. Annu Rev Genomics Hum Genet 15: 269-293.
- Heyn H, Moran S,
Hernando-Herraez I, et al. (2013) DNA methylation contributes to natural
human variation. Genome Res 23: 1363-1372.
- Iacobazzi V, Castegna A,
Infantino V, Andria G (2013) Mitochondrial DNA methylation as a
next-generation biomarker and diagnostic tool. Mol Genet Metab 110: 25-34.
- Kubota T, Takae H, Miyake K
(2012) Epigenetic mechanisms and therapeutic perspectives for
neurodevelopmental disorders. Pharmaceuticals (Basel) 25: 369-383.
- Cacabelos R (2014) Epigenomic
networking in drug development: from pathogenic mechanisms to
pharmacogenomics. Drug Dev Res 75: 348-365.
- Cacabelos R, Torrellas C
(2014) Epigenetic drug discovery for Alzheimer's disease. Expert Opin Drug
Discov 9: 1059-1086.
- Cacabelos R (2011) The path
to personalized medicine in mental disorders. In: Ritsner MS, editor. The
handbook of neuropsychiatric biomarkers, endophenotypes and genes, vol. 4.
Netherlands: Springer pp 3-63.
- Kang MG, Byun K, Kim JH, et
al. (2015) Proteogenomics of the human hippocampus: The road ahead.
Biochim Biophys Acta 1854: 788-797.
- Tognini P, Napoli D,
Pizzorusso T (2015) Dynamic DNA methylation in the brain: a new epigenetic
mark for experience-dependent plasticity. Front Cell Neurosci 9: 331
- Cacabelos
R, Cacabelos P, Torrellas C, et al. (2014) Pharmacogenomics of Alzheimer's
disease: novel therapeutic strategies for drug development. Methods Mol
Biol 1175: 323-556.
- Bestor TH, Edwards JR,
Boulard M (2015) Notes on the role of dynamic DNA methylation in mammalian
development. PNAS 112: 6796–6799.
- Gavery MR, Roberts SB (2013)
Predominant intragenic methylation is associated with gene expression
characteristics in a bivalve mollusc. PeerJ 1: 215.
- Wang J, Yu
JT, Tan MS, et al. (2013)
Epigenetic mechanisms in Alzheimer's disease: Implications for
pathogenesis and therapy. Ageing Res Rev 212: 1024-1041.
- Laurent L, Wong E, Li G, et
al. (2010) Dynamic changes in the human methylome during differentiation.
Genome Res 2: 320-331.
- Nebbioso A, Carafa V,
Benedetti R, Altucci L (2012) Trials with epigenetic drugs: An update. Mol
Oncol 6: 657-682.
- Cuadrado-Tejedor
M, Oyarzabal J, Lucas MP, et al. (2013) Epigenetic drugs in Alzheimer’s disease. BioMol
Concepts 4: 433-445.
- He Y, Ecker JR (2015) Non-CG
Methylation in the Human Genome. Annu Rev Genomics Hum Genet 16: 55-77.
- Konsoula Z, Barile FA (2012)
Epigenetic histone acetylation and deacetylation mechanisms in
experimental models of neurodegenerative disorders. J Pharmacol Toxicol
Methods 66: 215-220.
- Chouliaras L, Rutten BP,
Kenis G, et al. (2010) Epigenetic regulation in the pathophysiology of
Alzheimer's disease. Prog Neurobiol 90: 498-510.
- Clapier CR, Cairns BR (2009)
The biology of chromatin remodeling complexes. Ann Rev Biochem 78:
273-304.
- Huynh JL, Casaccia P (2013)
Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis
and treatment. Lancet Neurol 12: 195-206.
- Xu K, Dai XL, Huang HC, Jiang
ZF (2011) Targeting HDACs: a promising therapy for Alzheimer's disease.
Oxid Med Cell Longev.
- Gräff J, Tsai LH (2013) The
potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol
Toxicol 53: 311-330.
- Landgrave-Gómez J,
Mercado-Gómez O, Guevara-Guzmán R (2015) Epigenetic mechanisms in
neurological and neurodegenerative diseases. Front Cell Neurosci.
- ENCODE Project Consortium
(2012) An integrated encyclopedia of DNA elements in the human genome.
Nature 489: 57-74.
- Pennisi E (2012) Genomics.
ENCODE project writes eulogy for junk DNA. Science 337: 1159-1161.
- Ernst C, Morton CC (2013)
Identification and function of long non-coding RNA. Front Cell Neurosci 7:
168.
- Jiao AL, Slack FJ (2014)
RNA-mediated gene activation. Epigenetics 9: 27-36.
- Wu P, Zuo
X, Deng H, et al. (2013)
Roles of long noncoding RNAs in brain development, functional
diversification and neurodegenerative diseases. Brain Res Bull 97: 69-80.
- Li LC (2014) Chromatin
remodeling by the small RNA machinery in mammalian cells. Epigenetics 9:
45-52.
- Magistri M, Faghihi MA, St
Laurent G 3rd, Wahlestedt C (2012) Regulation of chromatin structure by
long noncoding RNAs: focus on natural antisense transcripts. Trends
Genet 28: 389-396.
- Velmeshev D, Magistri M,
Faghihi MA (2013) Expression of non-protein-coding antisense RNAs in
genomic regions related to autism spectrum disorders. Mol Autism 4: 32.
- Qureshi IA, Mehler MF (2010)
Genetic and epigenetic underpinnings of sex differences in the brain and
in neurological and psychiatric disease susceptibility. Prog Brain
Res 18: 77-95.
- Merelo V, Durand D,
Lescallette AR, et al. (2015) Associating schizophrenia, long non-coding
RNAs and neurostructural dynamics. Front Mol Neurosci 8: 57.
- Johnson R (2012) Long
non-coding RNAs in Huntington's disease neurodegeneration. Neurobiol
Dis 46: 245-254.
- Nishimoto Y, Nakagawa S,
Hirose T, et al. (2013) The long non-coding RNA nuclear-enriched abundant
transcript 1_2 induces paraspeckle formation in the motor neuron during
the early phase of amyotrophic lateral sclerosis. Mol Brain 6: 31.
- Messerschmidt DM, Knowles BB,
Solter D (2014) DNA methylation dynamics during epigenetic reprogramming
in the germline and preimplantation embryos. Genes Dev 28: 812-828.
- Verhoeven KJ, Preite V (2014)
Epigenetic variation in asexually reproducing organisms. Evolution 68:
644-655.
- McCarthy MM, Nugent BM (2015)
At the frontier of epigenetics of brain sex differences. Front Behav
Neurosci 9: 221.
- Cowan CS, Callaghan BL, Kan
JM, Richardson R (2015) The lasting impact of early-life adversity on
individuals and their descendants: Potential mechanisms and hope for
intervention. Genes Brain Behav.
- Irie M, Yoshikawa M, Ono R,
et al. (2015) Cognitive function related to the Sirh11/Zcchc16 gene
acquired from an LTR retrotransposon in Eutherians. PLoS Genet.
- Kraus TF, Kilinc S,
Steinmaurer M, et al. (2015) Profiling of methylation and demethylation
pathways during brain development and ageing. J Neural Transm (Vienna)
123: 189-203.
- Kinney SR, Pradhan S (2013)
Ten eleven translocation enzymes and 5-hydroxymethylation in mammalian
development and cancer. Adv Exp Med Biol 754: 57-79.
- Wang J, Tang J, Lai M, Zhang
H (2014) 5-Hydroxymethylcytosine and disease. Mutat Res Rev Mutat Res 762:
167-175.
- Fenoglio C, Ridolfi E,
Galimberti D, Scarpini E (2013) An emerging role for long non-coding RNA
dysregulation in neurological disorders. Int J Mol Sci 14: 20427-20442.
- Harony-Nicolas H, Mamrut S,
Brodsky L, et al. (2014) Brain region-specific methylation in the promoter
of the murine oxytocin receptor gene is involved in its expression
regulation. Psychoneuroendocrinology 39: 121-131.
- Rodrigues
HF, Souza TA, Ghiraldini FG, et al. (2014) Increased Age is Associated with
Epigenetic and Structural Changes in Chromatin from Neuronal Nuclei. J
Cell Biochem 115: 659-665.
- Zhao YQ, Jordan IK, Lunyak VV
(2013) Epigenetics components of aging in the central nervous system.
Neurotherapeutics 10: 647-663.
- Pan K, Chen Y, Roth M, et al.
(2013) HBP1-mediated transcriptional regulation of DNMT1 and its impact on
cell senescence. Mol Cell Biol 33: 887-903.
- Xu Z, Taylor JA (2014)
Genome-wide age-related DNA methylation changes in blood and other tissues
relate to histone modification, expression, and cancer. Carcinogenesis 35:
356-364.
- McClay JL, Aberg KA, Clark
SL, et al. (2014) A methylome-wide study of aging using massively parallel
sequencing of the methyl-CpG-enriched genomic fraction from blood in over
700 subjects. Hum Mol Genet 23: 1175-1185.
- Wagner M, Steinbacher J,
Kraus TF, et al. (2015) Age-dependent levels of 5-Methyl-,
5-Hydroxymethyl-, and 5-Formylcytosine in human and mouse brain tissues.
Angew Chem Int Ed Engl 54: 12511-12514.
- Nakamura A, Kawakami K,
Kametani F, et al. (2010) Biological significance of protein modifications
in aging and calorie restriction. Ann N Y Acad Sci 1197: 33-39.
- Wang CM, Tsai SN, Yew TW, et
al. (2010) Identification of histone methylation multiplicities patterns
in the brain of senescence-accelerated prone mouse 8.
Biogerontology 11: 87-102.
- Guarente L, Picard F (2005)
Calorie restriction--the SIR2 connection. Cell 120: 473-482.
- Haigis MC, Sinclair DA (2010)
Mammalian sirtuins: biological insights and disease relevance. Annu Rev
Pathol 5: 253-295.
- Raddatz G, Hagemann S, Aran
D, et al. (2013) Aging is associated with highly defined epigenetic
changes in the human epidermis. Epigenetics Chromatin 6: 36.
- Spallotta F, Cencioni C,
Straino S, et al. (2013) Enhancement of lysine acetylation accelerates
wound repair. Commun Integr Biol 6: 254-266.
- Hooten NN,
Abdelmohsen K, Gorospe M, et al. (2010) microRNA expression patterns reveal differential expression
of target genes with age. PLoS One 5: 10724.
- Hackl M, Brunner S,
Fortschegger K, et al. (2010) miR-17, miR-19b, miR-20a, and miR-106a are
down-regulated in human aging. Aging Cell 9: 291-296.
- Li N, Bates DJ, An J, et al.
(2011) Up-regulation of key microRNAs, and inverse down-regulation of
their predicted oxidative phosphorylationtarget genes, during aging in
mouse brain. Neurobiol Aging 32: 944-955.
- Morrison KE, Rodgers AB,
Morgan CP, Bale TL (2014) Epigenetic mechanisms in pubertal brain
maturation. Neuroscience 264: 17-24.
- Cacabelos R, Martínez-Bouza R
(2011) Genomics and pharmacogenomics of dementia. CNS Neurosci Ther 17:
566-576.
- Dalton VS, Kolshus E,
McLoughlin DM (2013) Epigenetics and depression: return of the repressed.
J Affect Disord 155: 1-12.
- Cacabelos R, Cacabelos P,
Aliev G (2013) Genomics and pharmacogenomics of antipsychotic drugs.
Open J Psychiatr 3: 46-139.
- Green MJ, Chia TY, Cairns MJ,
et al. (2014) Catechol-O-methyltransferase (COMT) genotype moderates the
effects of childhood trauma on cognition and symptoms in schizophrenia. J
Psychiatr Res 49: 43-50.
- Shirazi J, Shah S, Sagar D,
et al. (2013) Epigenetics, Drugs of Abuse, and the Retroviral Promoter. J
Neuroimmune Pharmacol 8: 1181-1196.
- Philibert RA, Beach SR, Lei
MK, Brody GH (2013) Changes in DNA methylation at the aryl hydrocarbon
receptor repressor may be a new biomarker for smoking. Clin Epigenetics 5:
19.
- Cacabelos R, Martínez-Bouza R
(2011) Genomics and pharmacogenomics of schizophrenia. CNS Neurosci
Ther 17: 541-565.
- Van den
Hove DL, Kompotis K, Lardenoije R, et al. (2014) Epigenetically regulated
microRNAs in Alzheimer's disease. Neurobiol Aging 35: 731-745.
- Rangasamy S, D'Mello SR,
Narayanan V (2013) Epigenetics, autism spectrum, and neurodevelopmental
disorders. Neurotherapeutics 10: 742-756.
- Kiser DP, Rivero O, Lesch KP
(2015) Annual research review: The (epi) genetics of neurodevelopmental
disorders in the era of whole-genome sequencing--unveiling the dark
matter. J Child Psychol Psychiatry 56: 278-295.
- Vallianatos CN, Iwase S
(2015) Disrupted intricacy of histone H3K4 methylation in
neurodevelopmental disorders. Epigenomics 7: 503-519.
- Kubota T, Miyake K, Hirasawa
T (2013) Role of epigenetics in Rett syndrome. Epigenomics 5: 583-592.
- Sáez MA,
Fernández-Rodríguez J, Moutinho C, et al. (2015) Mutations in JMJD1C are involved
in Rett syndrome and intellectual disability. Genet Med.
- Mbadiwe T, Millis RM (2015)
Epigenetics and autism. Autism Res Treat.
- Lahiri DK, Sokol DK, Erickson
C, et al. (2013) Autism as early neurodevelopmental disorder: evidence for
an sAPPα-mediated anabolic pathway. Front Cell Neurosci 7: 94.
- Loke YJ, Hannan AJ, Craig JM
(2015) The role of epigenetic change in autism spectrum disorders. Front Neurol
6: 107.
- Cacabelos R (2015) New
insights into the pathogenesis and pharmacogenomics of Attention Deficit
Hyperactivity Disorder. Metabolomics 5: 146.
- Cacabelos
R, Torrellas C, Tellado I, et al. (2015) Genomics, therapeutics and
pharmacogenomics of Attention-Deficit/Hyperactivity disorder: López-Muñoz
F, Álamo C, editors. Attention deficit hyperactivity disorder (ADHD):
Epidemiology, treatment and prevention. New York: Nova Science Publishers,
NY.
- Hall L, Kelley E (2014) The
contribution of epigenetics to understanding genetic factors in autism.
Autism 18: 872-881.
- Sullivan
JM, Badimon A, Schaefer U, et al. (2015) Autism-like syndrome is induced
by pharmacological suppression of BET proteins in young mice. J Exp Med
212: 1771-1781.
- Wang Y,
Zhao X, Ju W, et al. (2015) Genome-wide differential expression of synaptic long
noncoding RNAs in autism spectrum disorder. Transl Psychiatry 5: 660.
- Tabolacci E, Chiurazzi P
(2013) Epigenetics, fragile X syndrome and transcriptional therapy. Am J
Med Genet A 161: 2797-2808.
- Napoli E, Tassone F, Wong S,
et al. (2015) Mitochondrial citrate transporter-dependent metabolic
signature in the 22q11.2 deletion syndrome. J Biol Chem 290: 23240-23253.
- Vangeel EB, Izzi B, Hompes T,
et al. (2015) DNA methylation in imprinted genes IGF2 and GNASXL is
associated with prenatal maternal stress. Genes Brain Behav.
- Bartolomei MS (2009) Genomic
imprinting: employing and avoiding epigenetic processes. Genes De 23:
2124-2133.
- Girardot M, Feil R, Llères D (2013)
Epigenetic deregulation of genomic imprinting in humans: causal mechanisms
and clinical implications. Epigenomics 5: 715-728.
- Hannula-Jouppi K, Muurinen M,
Lipsanen-Nyman M, et al. (2014) Differentially methylated regions in
maternal and paternal uniparental disomy for chromosome 7. Epigenetics 9:
351-365.
- Matsubara
K, Kagami M, Nakabayashi K, et al. (2015) Exploration of hydroxymethylation
in Kagami-Ogata syndrome caused by hypermethylation of imprinting control
regions. Clin Epigenetics 7: 90.
- Abdolmaleky HM, Zhou JR,
Thiagalingam S (2015) An update on the epigenetics of psychotic diseases
and autism. Epigenomics 7: 427-449.
- López AJ, Wood MA (2015) Role
of nucleosome remodeling in neurodevelopmental and intellectual disability
disorders. Front Behav Neurosci 9: 100.
- Blaze J, Roth TL (2013)
Exposure to caregiver maltreatment alters expression levels of epigenetic
regulators in the medial prefrontal cortex. Int J Dev Neurosci 31:
804-810.
- Sullivan PF, Daly MJ,
O’Donovan M (2012) Genetic architectures of psychiatric disorders: the
emerging picture and its implications. Nat Rev Genet 13: 537–551.
- Ripke S, O’Dushlaine C,
Chambert K, et al. (2013) Genome-wide association analysis identifies 13
new risk loci for schizophrenia. Nat Genet 45: 1150–9.
100. Ibi D, González-Maeso J (2015) Epigenetic
signaling in schizophrenia. Cell Signal 27: 2131-2136.
101. Melka MG, Castellani CA, O'Reilly
R, Singh SM (2015) Insights into the origin of DNA methylation differences
between monozygotic twins discordant for schizophrenia. J Mol Psychiatry 3: 7.
102. Aberg KA, McClay JL, Nerella S, et
al. (2014) Methylome-wide association study of schizophrenia: identifying blood
biomarker signatures of environmental insults. JAMA Psychiatry 71: 255-264.
103. Pal M, Ebrahimi S, Oh G, et al.
(2015) High Precision DNA Modification Analysis of HCG9 in Major Psychosis.
104. Misiak B, Szmida
E, Karpiński P, et al. (2015)
Lower LINE-1 methylation in first-episode schizophrenia patients with the
history of childhood trauma. Epigenomics 27: 1-11.
105. Wockner LF, Noble EP, Lawford BR,
et al. (2014) Genome-wide DNA methylation analysis of human brain tissue from
schizophrenia patients. Transl Psychiatry 4: 339.
106. Shorter KR, Miller BH (2015)
Epigenetic mechanisms in schizophrenia. Prog Biophys Mol Biol 118: 1-7.
107. Kumar G, Clark SL, McClay JL, et
al. (2015) Refinement of schizophrenia GWAS loci using methylome-wide
association data. Hum Genet 134: 77-87.
108. Brucato N, DeLisi LE, Fisher SE,
Francks C (2014) Hypomethylation of the paternally inherited LRRTM1 promoter
linked to schizophrenia. Am J Med Genet B Neuropsychiatr Genet 165B: 555-563.
109. Kebir O,
Chaumette B, Fatjó-Vilas M, et al. (2014) Family-based association study of common variants, rare mutation
study and epistatic interaction detection in HDAC genes in schizophrenia.
Schizophr Res 160: 97-103.
110. Chase KA, Rosen C, Rubin LH, et
al. (2015) Evidence of a sex-dependent restrictive epigenome in schizophrenia.
J Psychiatr Res 65: 87-94.
111. Dong E, Ruzicka WB, Grayson DR,
Guidotti A (2015) DNA-methyltransferase1 (DNMT1) binding to CpG rich GABAergic
and BDNF promoters is increased in the brain of schizophrenia and bipolar
disorder patients. Schizophr Res 167: 35-41.
112. Mitchell AC, Jiang Y, Peter C,
Akbarian S (2015) Transcriptional regulation of GAD1 GABA synthesis gene in the
prefrontal cortex of subjects with schizophrenia. Schizophr Res 167: 28-34.
113. Gavin DP, Kusumo H, Sharma RP, et
al. (2015) Gadd45b and N-methyl-D-aspartate induced DNA demethylation in
postmitotic neurons. Epigenomics 7: 567-579.
114. Bator E, Latusz
J, Radaszkiewicz A, et al. (2015) Valproic acid (VPA) reduces sensorimotor gating deficits and
HDAC2 overexpression in the MAM animal model of schizophrenia. Pharmacol Rep
67: 1124-1129.
115. Viana J, Pidsley
R, Troakes C, et al. (2014)
Epigenomic and transcriptomic signatures of a Klinefelter syndrome (47, XXY)
karyotype in the brain. Epigenetics 9: 587-599.
116. Fisher HL, Murphy TM, Arseneault
L, et al. (2015) Methylomic analysis of monozygotic twins discordant for
childhood psychotic symptoms. Epigenetics 10: 1014-1023.
117. Xu J, He G, Zhu J, et al. (2014)
Prenatal nutritional deficiency reprogrammed postnatal gene expression in
mammal brains: implications for schizophrenia. Int J Neuropsychopharmacol.
118. Butler AA, Webb WM, Lubin FD
(2015) Regulatory RNAs and control of epigenetic mechanisms: expectations for
cognition and cognitive dysfunction. Epigenomics.
119. Kos A, Aschrafi A, Nadif Kasri N
(2015) The Multifarious Hippocampal Functions of MicroRNA-137. Neuroscientist.
120. Gottschalk MG, Wesseling H, Guest
PC, Bahn S (2014) Proteomic enrichment analysis of psychotic and affective
disorders reveals common signatures in presynaptic glutamatergic signaling and
energy metabolism. Int J Neuropsychopharmacol.
121. MacDonald ML, Ding Y, Newman J, et
al. (2015) Altered glutamate protein co-expression network topology linked to
spine loss in the auditory cortex of schizophrenia. Biol Psychiatry 77:
959-968.
122. Bayés À, Collins MO, Galtrey CM,
et al. (2014) Human post-mortem synapse proteome integrity screening for
proteomic studies of postsynaptic complexes. Mol Brain 7: 88.
123. Föcking M, Lopez LM, English JA,
et al. (2015) Proteomic and genomic evidence implicates the postsynaptic
density in schizophrenia. Mol Psychiatry 20: 424-432.
124. Wearne TA, Mirzaei M, Franklin JL,
et al. (2015) Methamphetamine-induced sensitization is associated with
alterations to the proteome of the prefrontal cortex: implications for the
maintenance of psychotic disorders. J Proteome Res 14: 397-410.
125. Ding YH, Guo JH, Hu QY, et al.
(2015) Protein Biomarkers in Serum of Patients with Schizophrenia. Cell Biochem
Biophys.
126. Al Awam K,
Haußleiter IS, Dudley E, et al. (2015) Multiplatform metabolome and proteome profiling identifies serum
metabolite and protein signatures as prospective biomarkers for schizophrenia.
J Neural Transm (Vienna) 122 Suppl 1: S111-22.
127. Iavarone F,
Melis M, Platania G, et al. (2014) Characterization of salivary proteins of schizophrenic and
bipolar disorder patients by top-down proteomics. J Proteomics 103: 15-22.
128. Saia-Cereda VM, Cassoli JS,
Schmitt A, et al. (2015) Proteomics of the corpus callosum unravel pivotal
players in the dysfunction of cell signaling, structure, and myelination in
schizophrenia brains. Eur Arch Psychiatry Clin Neurosci 265: 601-612.
129. Wesseling H, Want EJ, Guest PC, et
al. (2015) Hippocampal proteomic and metabonomic abnormalities in
neurotransmission, oxidative stress, and apoptotic pathways in a chronic
phencyclidine rat model. J Proteome Res 14: 3174-3187.
130. Schubert KO, Föcking M, Cotter DR
(2015) Proteomic pathway analysis of the hippocampus in schizophrenia and
bipolar affective disorder implicates 14-3-3 signaling, aryl hydrocarbon
receptor signaling, and glucose metabolism: Potential roles in GABAergic
interneuron pathology. Schizophr Res 167: 64-72.
131. English JA, Fan Y, Föcking M, et
al. (2015) Reduced protein synthesis in schizophrenia patient-derived olfactory
cells. Transl Psychiatry 5: 663.
132.
Januar
V, Saffery R, Ryan J (2015) Epigenetics and depressive disorders: a review of
current progress and future directions. Int J Epidemiol 44: 1364-1387.
133. Numata S, Ishii
K, Tajima A, et al. (2015)
Blood diagnostic biomarkers for major depressive disorder using multiplex DNA
methylation profiles: discovery and validation. Epigenetics 10: 135-141.
134. Kaut O, Schmitt I, Hofmann A, et
al. (2015) Aberrant NMDA receptor DNA methylation detected by epigenome-wide
analysis of hippocampus and prefrontal cortex in major depression. Eur Arch
Psychiatry Clin Neurosci 265: 331-341.
135. Sun H, Damez-Werno DM, Scobie KN,
et al. (2015) ACF chromatin-remodeling complex mediates stress-induced
depressive-like behavior. Nat Med 21: 1146-1153.
136. Choi S, Han KM,
Won E, et al. (2014)
Association of brain-derived neurotrophic factor DNA methylation and reduced
white matter integrity in the anterior corona radiata in major depression. J
Affect Disord 172C: 74-80.
137. Braithwaite EC, Kundakovic M,
Ramchandani PG, et al. (2015) Maternal prenatal depressive symptoms predict
infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 10: 408-417.
138. Januar V, Ancelin ML, Ritchie K,
et al. (2015) BDNF promoter methylation and genetic variation in late-life
depression. Transl Psychiatry 5: 619.
139.
Córdova-Palomera A, Fatjó-Vilas M, Palma-Gudiel H, et al.
(2015) Further
evidence of DEPDC7 DNA hypomethylation in depression: A study in adult twins. Eur Psychiatry
30: 715-718.
140. Córdova-Palomera
A, Fatjó-Vilas M, Gastó C, et al. (2015) Genome-wide methylation study on depression: differential
methylation and variable methylation in monozygotic twins. Transl Psychiatry 5:
557.
141. Gurnot C, Martin-Subero I, Mah SM,
et al. (2015) Prenatal antidepressant exposure associated with CYP2E1 DNA
methylation change in neonates. Epigenetics 10: 361-372.
142. Booij L, Szyf M, Carballedo A, et
al. (2015) DNA methylation of the serotonin transporter gene in peripheral
cells and stress-related changes in hippocampal volume: a study in depressed
patients and healthy controls. PLoS One 10: 0119061.
143. Frodl T, Szyf M, Carballedo A, et
al. (2015) DNA methylation of the serotonin transporter gene (SLC6A4) is
associated with brain function involved in processing emotional stimuli. J
Psychiatry Neurosci 40: 296-305.
144. Nghia NA, Hirasawa T, Kasai H, et
al. (2015) Long-term imipramine treatment increases N-methyl-d-aspartate
receptor activity and expression via epigenetic mechanisms. Eur J Pharmacol
752: 69-77.
145. Doh MS, Han DM,
Oh DH, et al. (2015)
Profiling of proteins regulated by venlafaxine during neural differentiation of
human cells. Psychiatry Investig 12: 81-91.
146. Stelzhammer V, Alsaif M, Chan MK,
et al. (2015) Distinct proteomic profiles in post-mortem pituitary glands from
bipolar disorder and major depressive disorder patients. J Psychiatr Res 60:
40-48.
147. Lee J, Joo EJ,
Lim HJ, et al. (2015)
Proteomic analysis of serum from patients with major depressive disorder to
compare their depressive and remission statuses. Psychiatry Investig 12:
249-259.
148. Shao WH, Chen JJ, Fan SH, et al.
(2015) Combined metabolomics and proteomics analysis of Major Depression in an
animal model: perturbed energy metabolism in the chronic mild stressed rat
cerebellum. OMICS 19: 383-392.
149. Wu D, Peng Y,
Zhou J, et al. (2015)
Identification and validation of argininosuccinate synthase as a candidate
urinary biomarker for major depressive disorder. Clin Chim Acta.
150. Zhan Y, Yang YT, You HM, et al.
(2014) Plasma-based proteomics reveals lipid metabolic and immunoregulatory
dysregulation in post-stroke depression. Eur Psychiatry 29: 307-315.
151. Cadet JL (2014) Epigenetics of
Stress, Addiction, and Resilience: Therapeutic Implications. Mol Neurobiol.
152. Yohn NL, Bartolomei MS, Blendy JA
(2015) Multigenerational and transgenerational inheritance of drug exposure:
The effects of alcohol, opiates, cocaine, marijuana, and nicotine. Prog Biophys
Mol Biol 118: 21-33.
153. Eiden LE, Weihe E (2011) VMAT2: a
dynamic regulator of brain monoaminergic neuronal function interacting with
drugs of abuse. Ann N Y Acad Sci 1216: 86-98.
154. Resendiz M, Chen Y, Oztürk NC,
Zhou FC (2013) Epigenetic medicine and fetal alcohol spectrum disorders.
Epigenomics 5: 73-86.
155. Jašarević E, Geary DC, Rosenfeld
CS (2012) Sexually selected traits: a fundamental framework for studies on
behavioral epigenetics. ILAR J 53: 253-269.
156. Pizzimenti CL, Lattal KM (2015)
Epigenetics and memory: causes, consequences and treatments for post-traumatic
stress disorder and addiction. Genes Brain Behav 14: 73-84.
157. Maddox SA, Schafe GE, Ressler KJ
(2013) Exploring Epigenetic Regulation of Fear Memory and Biomarkers Associated
with Post-Traumatic Stress Disorder. Front Psychiatry 4: 62.
158. Zovkic IB, Meadows JP, Kaas GA,
Sweatt JD (2013) Interindividual Variability in Stress Susceptibility: A Role
for Epigenetic Mechanisms in PTSD. Front Psychiatry 4: 60.
159. Laufer BI,
Kapalanga J, Castellani CA, et al. (2015) Associative DNA methylation changes in children with prenatal
alcohol exposure. Epigenomics.
160. Lardenoije R, Iatrou A, Kenis G,
et al. (2015) The epigenetics of aging and neurodegeneration. Prog Neurobiol
131: 21-64.
161. Cacabelos R,
Fernández-Novoa L, Lombardi V, et al. (2005) Molecular genetics of Alzheimer’s disease and aging. Methods
Find Exp Clin Pharmacol 27(Suppl A): 1-573.
162.
Takeda M, Martínez R, Kudo T, et al. (2010) Apolipoprotein E and
central nervous system disorders: Reviews of clinical findings. Psychiatry Clin
Neurosci 64: 592-607.
163.
Cacabelos R, Fernández-Novoa L, Martínez-Bouza R, et al. (2010) Future trends in the
pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6
variants. Pharmaceuticals 3: 3040-100.
164. Cacabelos R, Takeda M (2006)
Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease.
Drugs Future 31(Suppl B): 5-146.
165. Veerappan CS, Sleiman S, Coppola G
(2013) Epigenetics of Alzheimer's disease and frontotemporal dementia.
Neurotherapeutics 10: 709-721.
166. Mastroeni D,
Grover A, Delvaux E, et al. (2011) Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging
32: 1161-1180.
167. Mill J (2011) Toward an integrated
genetic and epigenetic approach to Alzheimer's disease. Neurobiol Aging 32:
1188-1191.
168. Edwards TM, Myers JP (2007)
Environmental exposures and gene regulation in disease etiology. Environ Health
Perspect 115: 1264-1270.
169. Nakagawa S, Kageyama Y (2014)
Nuclear lncRNAs as epigenetic regulators-Beyond skepticism. Biochim Biophys
Acta 1839: 215-222.
170. Hartley I, Elkhoury FF, Heon Shin
J, et al. (2013) Long-lasting changes in DNA methylation following short-term
hypoxic exposure in primary hippocampal neuronal cultures. PLoS One 8: 77859.
171. Liesz A, Zhou W, Na SY, et al.
(2013) Boosting regulatory T cells limits neuroinflammation in permanent cortical
stroke. J Neurosci 33: 17350-17362.
172. White AO, Wood MA (2014) Does
stress remove the HDAC brakes for the formation and persistence of long-term
memory? Neurobiol Learn Mem 112: 61-67.
173. Brochier C, Langley B (2013)
Chromatin modifications associated with DNA double-strand breaks repair as
potential targets for neurological diseases. Neurotherapeutics 10: 817-830.
174. Coppedè F, Migliore L (2010)
Evidence linking genetics, environment, and epigenetics to impaired DNA repair
in Alzheimer's disease. J Alzheimers Dis 20: 953-966.
175. Zawia NH, Lahiri DK,
Cardozo-Pelaez F (2009) Epigenetics, oxidative stress, and Alzheimer disease.
Free Radic Biol Med 46: 1241-1249.
176. Sultan FA, Sweatt JD (2013) The role
of the gadd45 family in the nervous system: a focus on neurodevelopment,
neuronal injury, and cognitive neuroepigenetics. Adv Exp Med Biol 793: 81-119.
177. Cacabelos R,
Torrellas C, López-Muñoz F (2014) Epigenomics of Alzheimer’s Disease. J Exp Clin Med 6: 75-82.
178. Hernández HG,
Mahecha MF, Mejía A, et al. (2014) Global long interspersed nuclear element 1 DNA methylation in a
Colombian sample of patients with Late-Onset al.zheimer's Disease. Am J
Alzheimers Dis Other Demen 29: 50-53.
179.
Bollati V, Galimberti D, Pergoli L, et al. (2011) DNA methylation in
repetitive elements and Alzheimer disease. Brain Behav Immun 25:
1078-1083.
180. Tohgi H, Utsugisawa K, Nagane Y,
et al. (1999) Reduction with age in methylcytosine in the promoter region -224
approximately-101 of the amyloid precursorprotein gene in autopsy human cortex.
Brain Res Mol Brain Res 70: 288-292.
181.
Octave JN, Pierrot N, Ferao Santos S, et al. (2013) From synaptic spines to
nuclear signaling: nuclear and synaptic actions of the amyloid precursor
protein. J Neurochem 126: 183-190.
182. Raina A, Kaul D (2010) LXR-α genomics programmes neuronal
death observed in Alzheimer's disease. Apoptosis 15: 1461-1469.
183. Coppedè F (2010) One-carbon
metabolism and Alzheimer's disease: focus on epigenetics. Curr Genomics 11:
246-260.
184. Wang SC, Oelze B, Schumacher A
(2008) Age-specific epigenetic drift in late-onset al.zheimer’s disease. PLoS
One 3: 2698.
185. Caesar I, Gandy S (2012) Evidence
that an APOE 4 ‘double whammy’ increases risk for Alzheimer’s disease. BMC
Med 10: 36.
186. Rauhala HE, Porkka KP, Saramäki
OR, et al. (2008) Clusterin is epigenetically regulated in prostate cancer. Int
J Cancer 123: 1601-1609.
187. Tohgi H, Utsugisawa K, Nagane Y,
et al. (1999) The methylation status of cytosines in a gene promoter region
alters with age to downregulate transcriptional activity in human cerebral
cortex. Neurosci Lett 275: 89-92.
188. Sontag E, Nunbhakdi-Craig V,
Sontag JM, et al. (2007) Protein phosphatase 2A methyltransferase links
homocysteine metabolism with tau and amyloid precursor protein regulation. J
Neurosci 27: 2751-2759.
189. Zhou XW, Gustafsson JÅ, Tanila H,
et al. (2008) Tau hyperphosphorylation correlates with reduced methylation of
protein phosphatase 2A. Neurobiol Dis 31: 386-394.
190.
Sanchez-Mut JV, Aso E, Panayotis N, et al. (2013) DNA methylation map of
mouse and human brain identifies target genes in Alzheimer's disease. Brain 136:
3018-3027.
191. Silva PN, Furuya
TK, Braga IL, et al. (2014)
Analysis of HSPA8 and HSPA9 mRNA expression and promoter methylation in the
brain and blood of Alzheimer's disease patients. J Alzheimers Dis 38: 165-170.
192. Chouliaras L,
Mastroeni D, Delvaux E, et al. (2013) Consistent decrease in global DNA methylation and
hydroxymethylation in the hippocampus of Alzheimer's disease patients.
Neurobiol Aging 34: 2091-2099.
193. Adwan L, Zawia NH (2013).
Epigenetics: a novel therapeutic approach for the treatment of Alzheimer's
disease. Pharmacol Ther 139: 41-50.
194. Zhang K, Schrag M, Crofton A, et
al. (2012) Targeted proteomics for quantification of histone acetylation in
Alzheimer’s disease. Proteomics 12: 1261-1268.
195. Ding H, Dolan PJ, Johnson GVW
(2008) Histone deacetylase 6 interacts with the microtubule-associated protein
tau. Neurochem 106: 2119-2130.
196. Govindarajan N, Rao
P, Burkhardt S, et al. (2013) Reducing HDAC6 ameliorates cognitive
deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med 5: 52–63.
197. Julien C, Tremblay C, Émond V
(2009) SIRT1 decrease parallels the accumulation of tau in alzheimer disease. J
Neuropathol Exp Neurol 68: 48-58.
198. Donmez G, Wang D, Cohen DE,
Guarente L (2010) SIRT1 suppresses-amyloid production by activating the
-secretase gene ADAM10. Cell 142: 320-332.
199. Ryu H, Barrup M, Kowall NW, McKee
AC (2008) P3-260: Epigenetic modification in a monozygotic twin with
Alzheimer’s disease. Alzheimers Dement 4: T598.
200. Ogawa O, Zhu X,
Lee HG, et al. (2003)
Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a
mitotic catastrophe? Acta Neuropathol 105: 524-528.
201. Myung NH, Zhu X, Kruman II (2008)
Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in
astrocytes. Age (Dordr) 30: 209-215.
202. Francis YI, Fà M, Ashraf H, et al.
(2009) Dysregulation of histone acetylation in the APP/PS1 mouse model of
Alzheimer’s disease. J Alzheimers Dis 18: 131-139.
203. Walker MP, LaFerla FM, Oddo SS,
Brewer GJ (2013) Reversible epigenetic histone modifications and Bdnf
expression in neurons with aging and from a mouse model of Alzheimer's disease.
Age (Dordr) 35: 519-531.
204. Liu R, Lei JX,
Luo C, et al. (2012)
Increased EID1 nuclear translocation impairs synaptic plasticity and memory
function associated with pathogenesis of Alzheimer's disease. Neurobiol Dis 45:
902-912.
205. Koshibu K, Gräff J, Beullens M, et
al. (2009) Protein phosphatase 1 regulates the histone code for long-term
memory. J Neurosci 29: 13079-13089.
206. Peleg S,
Sananbenesi F, Zovoilis A, et al. (2010) Altered histone acetylation is associated with age-dependent
memory impairment in mice. Science 328: 753-756.
207. Fischer A, Sananbenesi F, Wang X,
et al. (2007) Recovery of learning and memory is associated with chromatin
remodelling. Nature 447: 178-182.
208. Guan
JS, Haggarty SJ, Giacometti E, et al. (2009) HDAC2 negatively regulates memory
formation and synaptic plasticity. Nature 459: 55-60.
209. Gräff
J, Rei D, Guan JS, et al. (2012) An epigenetic blockade of cognitive functions in the
neurodegenerating brain. Nature 483: 222-226
210. Kim MS, Akhtar
MW, Adachi M, et al. (2012) An essential role for histone deacetylase 4 in
synaptic plasticity and memory formation. J Neurosci 32: 10879-10886.
211. Ishimaru N, Fukuchi
M, Hirai A, et al. (2010) Differential epigenetic regulation of BDNF and
NT-3 genes by trichostatin A and 5-aza-2’-deoxycytidine in Neuro-2a cells.
Biochem Biophys Res Commun 394: 173-177.
212. Tian F, Marini
AM, Lipsky RH (2010) Effects of histone deacetylase inhibitor Trichostatin
A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by
rat hippocampal neurons. Ann N Y Acad Sci 1199: 186-193.
213. Gupta S, Kim SY, Artis
S, et al. (2010) Histone methylation regulates memory formation. J Neurosci 30:
3589-3599.
214. Lithner CU, Hernandez CM, Nordberg
A, Sweatt JD (2009) Epigenetic changesrelated to beta-amyloid-implications for
Alzheimer’s disease. Alzheimers Dement 5: 304.
215. Vogel-Ciernia A, Wood MA (2014)
Neuron-specific chromatin remodeling: A missing link in epigenetic mechanisms
underlying synaptic plasticity, memory, and intellectual disability disorders.
Neuropharmacol 80: 18-27.
216. Zhang L, Zhang Y, Chou CJ, et al.
(2014) Histone deacetylase inhibitors with enhanced enzymatic inhibition
effects and potent in vitro and in vivo antitumor activities. Chem Med Chem 9:
638-648.
217. Pirooznia SK, Elefant F (2013) A
HAT for sleep?: epigenetic regulation of sleep by Tip60 in Drosophila. Fly
(Austin) 7: 99-104.
218. Aljada A, Dong L, Mousa SA (2010)
Sirtuin-targeting drugs: Mechanisms of action and potential therapeutic
applications. Curr Opin Investig Drugs 11: 1158-1168.
219. Oey NE, Leung
HW, Ezhilarasan R, et al. (2015) A Neuronal Activity-Dependent Dual Function Chromatin-Modifying
Complex Regulates Arc Expression (1,2,3). Neuro.
220. Kumar P, Dezso
Z, Mackenzie C, et al. (2013)
Circulating miRNA biomarkers for Alzheimer's disease. PLoS One.
221. Leidinger P, Backes C, Deutscher
S, et al. (2013) A blood based 12-miRNA signature of Alzheimer disease
patients. Genome Biol 14: 78.
222. Alexandrov PN, Dua P, Hill JM, et
al. (2012) microRNA (miRNA) speciation in Alzheimer's disease (AD)
cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int J Biochem Mol Biol
3: 365-373.
223. Qureshi IA, Mehler MF (2011)
Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr
Neurol Neurosci Rep 11: 464-473.
224. Enciu AM, Popescu BO,
Gheorghisan-Galateanu A (2012) MicroRNAs in brain development and degeneration.
Mol Biol Rep 39: 2243-2252.
225. Mallick B, Ghosh Z (2011) A complex
crosstalk between polymorphic microRNA target sites and AD prognosis. RNA Biol
8: 665-673.
226. Long, JM, Lahiri DK (2011)
MicroRNA-101 downregulates Alzheimer’s amyloid- precursor protein levels in
human cell cultures and is differentially expressed. Biochem Biophys Res
Commun 404: 889-895.
227. Liu W, Liu
C, Zhu J, et al. (2012)
MicroRNA-16 targets amyloid precursor protein to potentially modulate
Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging 33: 522-534.
228. Smith P, Al Hashimi A, Girard J,
et al. (2011) In vivo regulation of amyloid precursor protein neuronal splicing
by microRNAs. J Neurochem 116: 240-247.
229. Fang M, Wang J, Zhang X
(2012) The miR-124 regulates the expression of BACE1/-secretase correlated with
cell death in Alzheimer’s disease. Toxicol Lett 209: 94-105.
230. Hébert
SS, Horré K, Nicolaï L, et al. (2008) Loss of microRNA cluster miR-29a/b-1
in sporadic Alzheimer’s disease correlates with increased BACE1/-secretase
expression. Proc Natl Acad Sci USA 105: 6415-6420.
231. Zong Y, Wang H, Dong W,
et al. (2011) miR-29c regulates BACE1 protein expression. Brain Res 1395:
108-115.
232. Wang WX, Rajeev BW, Stromberg AJ,
et al. (2008) The expression of microRNA miR-107 decreases early in Alzheimer’s
disease and may accelerate disease progression through regulation of -site
amyloid precursor protein-cleaving enzyme 1. J Neurosci 28: 1213-1223.
233. Boissonneault V, Plante
I, Rivest S, Provost P (2009) MicroRNA-298 and microRNA-328 regulate
expression of mouse -amyloid precursor protein-converting enzyme 1. J Biol Chem
284: 1971-1981.
234. Zhu HC, Wang LM, Wang M,
et al. (2012) MicroRNA-195 downregulates Alzheimer’s disease amyloid-
production by targeting BACE1. Brain Res Bull 88: 596-601.
235. Faghihi MA, Modarresi
F, Khalil AM, et al. (2008) Expression of a noncoding RNA is elevated in
Alzheimer’s disease and drives rapid feed-forward regulation of -secretase. Nat
Med 14: 723-730.
236. Faghihi MA, Zhang
M, Huang J, et al. (2010) Research evidence for natural antisense
transcript-mediated inhibition of microRNA function. Genome Biol 11: R56.
237. Massone
S, Ciarlo E, Vella S, et al. (2012) NDM29, A RNA polymerase III-dependent
non coding RNA, promotes amyloidogenic processing of amyloid precursor protein
(APP) and amyloid secretion. Biochim Biophys Acta 1823: 1170-1177.
238. Wang
WX, Huang Q, Hu Y, et al. (2011) Patterns of microRNA expression in normal and early Alzheimer’s
disease human temporal cortex: white matter versus gray matter. Acta
Neuropathol 121: 193-205.
239. Wang WX, Wilfred BR, Madathil SK,
et al. (2010) miR-107 regulates granulin/progranulin with implications for
traumatic brain injury and neurodegenerative disease. Am J Pathol 177: 334-345.
240. Geekiyanage H, Chan C (2011)
MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid,
novel targets in sporadic Alzheimer’s Disease. J Neurosci 31: 14820-14830.
241. Akram A, Schmeidler
J, Katsel P, et al. (2010) Increased expression of cholesterol transporter
ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain
Res 1318: 167-177.
242. Massone S, Vassallo I, Fiorino G,
et al. (2011) 17A, a novel non-coding RNA, regulates GABA B alternative
splicing and signaling in response to inflammatory stimuli and in Alzheimer
disease. Neurobiol Dis 41: 308-317.
243. Smith PY, Delay
C, Girard J, et al. (2011) MicroRNA-132 loss is associated with tau exon
10 inclusion in progressive supranuclear palsy. Hum Mol Genet 20: 4016-4024.
244. Hébert SS, Papadopoulou
AS, Smith P, et al. (2010) Genetic ablation of Dicer in adult forebrain
neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum
Mol Genet 19: 3959-3969.
245. Caputo
V, Sinibaldi L, Fiorentino A, et al. (2011) Brain derived neurotrophic factor
(BDNF) expression is regulated by microRNAs miR-26a and miR-26b allele-specific
binding. PLoS One 6: 28656.
246. Mohamed JS, Lopez
MA, Boriek AM (2010) Mechanical stretch up-regulates microRNA-26a and
induces human airway smooth muscle hypertrophy by suppressing glycogen synthase
kinase 3. J Biol Chem 285: 29336-29347.
247. Cogswell JP, Ward
J, Taylor IA, et al. (2008) Identification of miRNA changes in Alzheimer’s
disease brain and CSF yields putative biomarkers and insights into disease
pathways. J Alzheimers Dis 14: 27-41.
248. Bilen J, Liu N, Burnett
BG, et al. (2006) MicroRNA pathways modulate polyglutamine-induced
neurodegeneration. Mol Cell 24: 157-163.
249. Min SW, Cho
SH, Zhou Y, et al. (2010)
Acetylation of tau inhibits its degradation and contributes to tauopathy.
Neuron 67: 953-966.
250. Schonrock N, Humphreys
DT, Preiss T, Götz J (2012) Target gene repression mediated by miRNAs
miR-181c and miR-9 both of which are down-regulatedby amyloid-. J Mol Neurosci
46: 324-335.
251. Zovoilis A, Agbemenyah
HY, Agis-Balboa RC, et al. (2011) microRNA-34c is a novel target to treat
dementias. EMBO J 30: 4299-4308.
252. Carrettiero DC, Hernandez
I, Neveu P, et al. (2009) The cochaperone BAG2 sweeps paired helical
filament-insoluble tau from the microtubule. J Neurosci 29: 2151-2161.
253. Cui JG, Li
YY, Zhao Y, et al. (2010)
Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1)
and IRAK-2 by microRNA-146a and NF-(B in stressed human astroglial cells and in
Alzheimer disease. J Biol Chem 285: 38951-38960.
254. Taganov KD, Boldin
MP, Chang KJ, Baltimore D (2006) NF-B-dependent induction of microRNA
miR-146, an inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA 103: 12481-12486.
255. Lukiw WJ, Zhao Y, Cui JG
(2008) An NF-(B-sensitive micro RNA-146a-mediated inflammatory circuit in
alzheimer disease and in stressed human brain cells.J Biol Chem 283:
31315-31322.
256. Li YY, Cui JG,
Dua P, et al. (2011)
Differential expression of miRNA-146a-regulated inflammatory genes in human
primary neural, astroglial and microglial cells. Neurosci Lett 499: 109-113.
257. Sethi P, Lukiw WJ (2009) Micro-RNA
abundance and stability in human brain: specific alterations in Alzheimer’s
disease temporal lobe neocortex. Neurosci Lett 459: 100-104.
258. Popov N, Gil J (2010) Epigenetic
regulation of the INK4b-ARF-INK4a locus: in sickness and in health.Epigenetics
25: 685-690.
259. Blennow K, Hampel H, Weiner M,
Zetterberg H (2010) Cerebrospinal fluid and plasma biomarkers in Alzheimer
disease. Nat Rev Neurol 6: 131-144.
260. Ahmed RM, Paterson RW, Warren JD,
et al. (2014) Biomarkers in dementia: clinical utility and new directions. J
Neurol Neurosurg Psychiatry 85: 1426–1434.
261.
Chiam
JT, Dobson RJ, Kiddle SJ, Sattlecker M (2015) Are blood-based protein
biomarkers for Alzheimer's disease also involved in other brain disorders? A systematic
review. J Alzheimers Dis 43: 303-314.
262. Shah DJ, Rohlfing F, Anand S, et
al. (2015) Discovery and subsequent confirmation of novel serum biomarkers
diagnosing Alzheimer's disease. J Alzheimers Dis.
263. Kiddle SJ, Steves CJ, Mehta M, et
al. (2015) Plasma protein biomarkers of Alzheimer's disease endophenotypes in
asymptomatic older twins: early cognitive decline and regional brain volumes.
Transl Psychiatry 5: 584.
264. Hendrickson RC, Lee AY, Song Q, et
al. (2015) High resolution discovery proteomics reveals candidate disease
progression markers of Alzheimer's disease in human Cerebrospinal Fluid. PLoS
One 10: 0135365.
265.
Gentier RJ, Verheijen BM, Zamboni M, et al. (2015) Localization of mutant
ubiquitin in the brain of a transgenic mouse line with proteasomal inhibition
and its validation at specific sites in Alzheimer's disease. Front Neuroanat
9: 26.
266. Bruggink KA, Kuiperij HB, Gloerich
J, et al. (2015) Dickkopf-related protein 3 is a potential Aβ-associated protein in Alzheimer's
Disease. J Neurochem 134: 1152-1162.
267.
Palmigiano A, Barone R, Sturiale L, et al. (2015) CSF N-glycoproteomics for
early diagnosis in Alzheimer's disease. J Proteomics.
268.
Drummond
ES, Nayak S, Ueberheide B, Wisniewski T (2015) Proteomic analysis of neurons
microdissected from formalin-fixed, paraffin-embedded Alzheimer's disease brain
tissue. Sci Rep 5: 15456.
269. Triplett JC, Swomley AM, Cai J, et
al. (2015) Quantitative Phosphoproteomic Analyses of the Inferior Parietal
Lobule from Three Different Pathological Stages of Alzheimer's Disease. J
Alzheimers Dis.
270.
Anderson
KW, Turko IV (2015) Histone post-translational modifications in frontal cortex
from human donors with Alzheimer's disease. Clin Proteomics 12: 26.
271.
Fu Y, Zhao D, Pan B, et al. (2015) Proteomic analysis of protein
expression throughout disease progression in a mouse model of Alzheimer's
Disease. J Alzheimers Dis 47: 915-926.
272. Shen L, Chen C, Yang A, et al.
(2015) Redox proteomics identification of specifically carbonylated proteins in
the hippocampi of triple transgenic Alzheimer's disease mice at its earliest
pathological stage. J Proteomics 123: 101-113.
273. Bai B, Hales CM, Chen PC, et al.
(2013) U1 small nuclear ribonucleoprotein complex and RNA splicing alterations
in Alzheimer's disease. Proc Natl Acad Sci U S A 110: 16562-16567.
274.
Ly
H, Despa F (2015) Hyperamylinemia as a risk factor for accelerated cognitive
decline in diabetes. Expert Rev Proteomics.
275. Gunawardana CG, Mehrabian M, Wang
X, et al. (2015) The human tau interactome: binding to the ribonucleoproteome,
and impaired binding of the P301L mutant to chaperones and the proteasome. Mol
Cell Proteomics.
276.
Mitra
G, Gupta S, Poddar A, Bhattacharyya B (2015) MAP2c prevents arachidonic
acid-induced fibril formation of tau: Role of chaperone activity and
phosphorylation. Biophys Chem 205: 16-23.
277. Derisbourg M, Leghay C, Chiappetta
G, et al. (2015) Role of the Tau N-terminal region in microtubule stabilization
revealed by new endogenous truncated forms. Sci Rep 5: 9659.
278.
Wang
C, Zhao J, Xu R, et al. (2015) Identification of pivotal markers in vascular
dementia based on proteomics data. Dement Geriatr Cogn Disord. 39: 312-320.
279.
Feng
Y, Jankovic J, Wu YC (2015) Epigenetic mechanisms in Parkinson's disease. J Neurol Sci
349: 3-9.
280.
Nuytemans
K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson
disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2
genes: a mutation update. Hum Mutat 31: 763-780.
281.
Coppedè
F (2012) Genetics and epigenetics of Parkinson’s disease. Scientific World J.
282.
imón-Sánchez
J, Heutink P, Gasser T (2015) International Parkinson's Disease Genomics
Consortium (IPDGC). Variation in PARK10 is not associated with risk and age at
onset of Parkinson's disease in large clinical cohorts. Neurobiol Aging.
283. McGough IJ, Steinberg F, Jia D, et
al. (2014) Retromer binding to FAM21 and the WASH complex is perturbed by the
Parkinson disease-linked VPS35 (D620N) mutation. Curr Biol 24: 1670-1676.
284. Ozgul S, Kasap M, Akpinar G, et
al. (2015) Linking a compound-heterozygous Parkin mutant (Q311R and A371T) to
Parkinson's disease by using proteomic and molecular approaches. Neurochem Int
1: 1385-1386.
285.
Pihlstrøm
L, Berge V, Rengmark A, Toft M (2015) Parkinson's disease correlates with
promoter methylation in the α-synuclein gene. Mov Disord 30: 577-580.
286.
Ammal
Kaidery N, Tarannum S, Thomas B (2013) Epigenetic landscape of Parkinson’s
disease: emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics
10: 698-708.
287.
Jowaed
A, Schmitt I, Kaut O, Wüllner U (2010) Methylation regulates alpha-synuclein
expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci 30:
6355–6359.
288. Desplats P, Spencer B, Coffee E,
et al. (2011) Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel
mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem 286:
9031–9037.
289.
Masliah
E, Dumaop W, Galasko D, Desplats P (2013) Distinctive patterns of DNA
methylation associated with Parkinson disease: identification of concordant
epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8:
1030-1038.
290.
Moore
K, McKnight AJ, Craig D, O'Neill F (2014) Epigenome-wide association study for
Parkinson's disease. Neuromolecular Med 16: 845-855.
291.
Kolosova
NG, Shcheglova TV, Sergeeva SV, Loskutova LV (2006) Long-term antioxidant
supplementation attenuates oxidative stress markers and cognitive deficits in
senescent-accelerated OXYS rats. Neurobiol Aging 27: 1289-1297.
292.
Kontopoulos
E, Parvin JD, Feany MB (2006) Alpha-synuclein acts in the nucleus to inhibit
histone acetylation and promote neurotoxicity. Hum Mol Genet 15:
3012-3023.
293. Wu R, Chen H, Ma J, et al. (2015)
c-Abl-p38α signaling plays
an important role in MPTP-induced neuronal death. Cell Death Differ.
294. Jin H, Kanthasamy A, Harischandra
DS, et al. (2014) Histone hyperacetylation up-regulates protein kinase Cδ in dopaminergic neurons to induce
cell death: relevance to epigenetic mechanisms of neurodegeneration in
Parkinson disease. J Biol Chem 89: 34743-34767.
295.
Reyniers L, Del Giudice MG, Civiero L, et al. (2014) Differential
protein-protein interactions of LRRK1 and LRRK2 indicate roles in distinct
cellular signaling pathways. J Neurochem 131: 239-250.
296.
Luerman GC, Nguyen C, Samaroo H, et al. (2014) Phosphoproteomic evaluation
of pharmacological inhibition of leucine-rich repeat kinase 2 reveals
significant off-target effects of LRRK-2-IN-1. J Neurochem 128:
561-576.
297.
Tang Y, Li T, Li J, et al. (2014) Jmjd3 is essential for the epigenetic
modulation of microglia phenotypes in the immune pathogenesis of Parkinson's
disease. Cell Death Differ 21: 369-380.
298. Kabaria S, Choi DC, Chaudhuri AD,
et al. (2015) MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression.
Free Radic Biol Med 89: 548-556.
299.
Niere F, Namjoshi S, Song E, et al. (2015) Analysis of proteins that
rapidly change upon mTORC1 repression identifies PARK7 as a novel protein
aberrantly expressed in Tuberous Sclerosis Complex. Mol Cell Proteomics.
300.
Liu Y, Zhou Q, Tang M, et al. (2015) Upregulation of alphaB-crystallin
expression in the substantia nigra of patients with Parkinson's disease. Neurobiol Aging
36: 1686-1691.
301.
Riley
BE, Gardai SJ, Emig-Agius D, et al. (2014) Systems-based analyses of brain
regions functionally impacted in Parkinson's disease reveals underlying causal
mechanisms. PLoS One 9: 102909.
302. Triplett JC, Zhang Z, Sultana R,
et al. (2015) Quantitative expression proteomics and phosphoproteomics profile
of brain from PINK1 knockout mice: insights into mechanisms of familial
Parkinson's disease. J Neurochem 133: 750-765.
303.
Ordureau A, Sarraf SA, Duda DM, et al. (2014) Quantitative proteomics
reveal a feedforward mechanism for mitochondrial PARKIN translocation and
ubiquitin chain synthesis. Mol Cell 56: 360-375.
304.
Choi
DC, Chae YJ, Kabaria S, et al. (2014) MicroRNA-7 protects against
1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J Neurosci 34:
12725-12737.
305.
Wang KZ, Zhu J, Dagda RK, et al. (2014) ERK-mediated phosphorylation of TFAM
downregulates mitochondrial transcription: implications for Parkinson's
disease. Mitochondrion 17: 132-140.
306. Shin DI, Oh YJ (2014) Tumor
necrosis factor-associated protein 1 (TRAP1) is released from the mitochondria
following 6-hydroxydopamine treatment. Exp Neurobiol 23: 65-76.
307.
Lin
X, Shi M, Masilamoni JG, et al. (2015) Proteomic profiling in MPTP monkey model
for early Parkinson disease biomarker discovery. Biochim Biophys Acta
1854: 779-787.
308.
Rakshit
H, Rathi N, Roy D (2014) Construction and analysis of the protein-protein interaction
networks based on gene expression profiles of Parkinson's disease. PLoS One 9:
103047.
309.
Chahine
LM, Stern MB, Chen-Plotkin A (2014) Blood-based biomarkers for Parkinson's
disease. Parkinsonism Relat Disord 20: 99-103.
310. Shi M, Movius J, Dator R, et al.
(2015) Cerebrospinal fluid peptides as potential Parkinson disease biomarkers:
a staged pipeline for discovery and validation. Mol Cell Proteomics 14:
544-555.
311.
Licker V, Turck N, Kövari E, et al. (2014) Proteomic analysis of human
substantia nigra identifies novel candidates involved in Parkinson's disease
pathogenesis. Proteomics 14: 784-794.
312.
Orr
HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:
575–621.
313.
Scaramuzzino
C, Casci I, Parodi S, et al. (2015) Protein arginine methyltransferase 6
enhances polyglutamine-expanded androgen receptor function and toxicity in
spinal and bulbar muscular atrophy. Neuron 85: 88-100.
314.
Lee
J, Hwang YJ, Kim KY (2013) Epigenetic Mechanisms of Neurodegeneration in
Huntington's Disease. Neurotherapeutics 10: 664-676.
315.
Moumné
L, Betuing S, Caboche J (2013) Multiple Aspects of Gene Dysregulation in
Huntington's Disease. Front Neurol 4: 127.
316.
Laffita-Mesa JM, Bauer PO, Kourí V, et al. (2012) Epigenetics DNA methylation
in the core ataxin-2 gene promoter: novel physiological and pathological
implications. Hum Genet 131: 625-638.
317.
Kipkorir
T, Colangelo CM, Manuelidis L (2015) Proteomic analysis of host brain
components that bind to infectious particles in Creutzfeldt-Jakob disease. Proteomics 15:
2983-2998.
318. Oeckl P, Steinacker P, Feneberg E,
Otto M (2015) Cerebrospinal fluid proteomics and protein biomarkers in
frontotemporal lobar degeneration: Current status and future perspectives.
Biochim Biophys Acta 1854: 757-768.
319. Ye C, Li L (2014)
5-hydroxymethylcytosine: A new insight into epigenetics in cancer. Cancer Biol
Ther 15: 10-15.
320. Wahlin KJ, Enke RA, Fuller JA, et
al. (2013) Epigenetics and cell death: DNA hypermethylation in programmed
retinal cell death. PLoS One 8: 79140.
321. Kraus TF, Greiner A, Steinmaurer M,
et al. (2015) Genetic Characterization of Ten-Eleven-Translocation
Methylcytosine Dioxygenase Alterations in Human Glioma. J Cancer 6: 832-842.
322. Choudhury SR, Cui Y, Milton JR, et
al. (2015) Selective increase in subtelomeric DNA methylation: an epigenetic
biomarker for malignant glioma. Clin Epigenetics 7: 107.
323. Li G, Warden C, Zou Z, et al.
(2013) Altered expression of polycomb group genes in glioblastoma multiforme.
PLoS One 8: 80970.
324. Salhia B, Kiefer J, Ross JT, et
al. (2014) Integrated genomic and epigenomic analysis of breast cancer brain
metastasis. PLoS One 9: 85448.
325. Nguyen KV (2015) Epigenetic
Regulation in Amyloid Precursor Protein with Genomic Rearrangements and the
Lesch-Nyhan Syndrome. Nucleosides Nucleotides Nucleic Acids 34: 674-690.
326. Yang H, Lee SM, Gao B, et al.
(2013) The histone deacetylase Sirtuin 1 deacetylates IRF1 and programs
dendritic cells to control Th17 differentiation during autoimmune inflammation.
J Biol Chem 2288: 37256-37266.
327. Shukla S, Shankavaram UT, Nguyen
P, et al. (2015) Radiation-Induced Alteration of the Brain Proteome:
Understanding the Role of the Sirtuin 2 Deacetylase in a Murine Model. J
Proteome Res 14: 4104-4117.
328. Kiguchi N, Kobayashi Y, Saika F,
Kishioka S (2013) Epigenetic upregulation of CCL2 and CCL3 via histone
modifications in infiltrating macrophages after peripheral nerve injury.
Cytokine 64: 666-672.
329. Küçükali Cİ,
Kürtüncü M, Çoban A, et al. (2015) Epigenetics of multiple sclerosis: an updated review.
Neuromolecular Med 17: 83-96.
330. van den Elsen
PJ, van Eggermond MC, Puentes F, et al. (2014) The epigenetics of multiple sclerosis
and other related disorders. Mult Scler Relat Disord 3: 163-175.
331. Koch MW, Metz LM, Kovalchuk O
(2013) Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol
9: 35-43.
332. Eising E, A Datson N, van den
Maagdenberg AM, Ferrari MD (2013) Epigenetic mechanisms in migraine: a
promising avenue? BMC Med 11: 26.
333. Williams-Karnesky RL, Sandau US,
Lusardi TA, et al. (2013) Epigenetic changes induced by adenosine augmentation
therapy prevent epileptogenesis. J Clin Invest 123: 3552-3563.
334. Jones T, King OD, Himeda CL, et
al. (2015) Individual epigenetic status of the pathogenic D4Z4 macrosatellite
correlates with disease in facioscapulohumeral muscular dystrophy. Clin
Epigenetics 7: 37.
335. Sacconi S, Salviati L, Desnuelle C
(2015) Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 1852:
607-614.
336. Daxinger L, Tapscott SJ, van der
Maarel SM (2015) Genetic and epigenetic contributors to FSHD. Curr Opin Genet
Dev 33: 56-61.
337. Consalvi S, Saccone V, Mozzetta C
(2014) Histone deacetylase inhibitors: a potential epigenetic treatment for
Duchenne muscular dystrophy. Epigenomics 6: 547-560.
338. Lessans S, Dorsey SG (2013) The
role for epigenetic modifications in pain and analgesia response. Nurs Res
Pract 2013: 961493.
339. Descalzi G,
Ikegami D, Ushijima T, et al. (2015) Epigenetic mechanisms of chronic pain. Trends Neurosci 38:
237-246.
340. Aliper AM, Csoka AB, Buzdin A, et
al. (2015) Signaling pathway activation drift during aging: Hutchinson-Gilford
Progeria Syndrome fibroblasts are comparable to normal middle-age and old-age
cells. Aging (Albany NY) 7: 26-37.
341. Arancio W,
Pizzolanti G, Genovese SI, et al. (2014) Epigenetic involvement in Hutchinson-Gilford progeria syndrome:
a mini-review. Gerontology 60: 197-203.
342. Gabriel D, Roedl D, Gordon LB,
Djabali K (2015) Sulforaphane enhances progerin clearance in Hutchinson-Gilford
progeria fibroblasts. Aging Cell 14: 78-91.
343. Luzhna L, Kathiria P, Kovalchuk O
(2013) Micronuclei in genotoxicity assessment: from genetics to epigenetics and
beyond. Front Genet 24: 131.
344. Sen N, Kumari R, Singh MI, Das S
(2013) HDAC5, a key component in temporal regulation of p53-mediated
transactivation in response to genotoxic stress. Mol Cell 52: 406-420.
345. Mack SC, Witt H, Piro RM, et al.
(2014) Epigenomic alterations define lethal CIMP-positive ependymomas of
infancy. Nature 506: 445-450.
346. Krishna SM, Trollope AF, Golledge
J (2015) The relevance of epigenetics to occlusive cerebral and peripheral
arterial disease. Clin Sci (Lond) 128: 537-558.
347. Shimamura M, Nakagami H, Osako MK,
et al. (2014) OPG/RANKL/RANK axis is a critical inflammatory signaling system
in ischemic brain in mice. Proc Natl Acad Sci USA 111: 8191-8196.
348. Vermulst M, Denney AS, Lang MJ, et
al. (2015) Transcription errors induce proteotoxic stress and shorten cellular
lifespan. Nat Commun 6: 8065.
349. Cacabelos R, Torrellas C (2015)
Pharmacogenomics of Antidepressants. J Psychiatr Depress Anxiety 1: 001.
350. Del Campo M,
Jongbloed W, Twaalfhoven HA, et al. (2015) Facilitating the validation of novel protein biomarkers for
dementia: An optimal workflow for the development of sandwich immunoassays.
Front Neurol 6: 202.
351. Shevchenko G, Konzer A, Musunuri
S, Bergquist J (2015) Neuroproteomics tools in clinical practice. Biochim
Biophys Acta 1854: 705-717.
352. Spellman DS, Wildsmith KR, Honigberg
LA, et al. (2015) Development and evaluation of a multiplexed mass spectrometry
based assay for measuring candidate peptide biomarkers in Alzheimer's Disease
Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl 9: 715-731.
353. Lista S, O'Bryant SE, Blennow K,
et al. (2015) Biomarkers in Sporadic and Familial Alzheimer's Disease. J
Alzheimers Dis 47: 291-317.
354. Harrison IF, Dexter DT (2013)
Epigenetic targeting of histone deacetylase: therapeutic potential in
Parkinson's disease? Pharmacol Ther 140: 34-52.
355. Rasool M, Malik
A, Naseer MI, et al. (2015)
The role of epigenetics in personalized medicine: challenges and opportunities.
BMC Med Genomics 8: 5.
356. Monte AA, Brocker C, Nebert DW, et
al. (2014) Improved drug therapy:
triangulating phenomics with genomics and metabolomics. Hum Genomics 8: 16.
357. Siggens L, Ekwall K (2014)
Epigenetics, chromatin and genome organization: recent advances from the ENCODE
project. J Intern Med 276: 201-214.
358. Kundakovic M, Champagne FA (2015)
Early-life experience, epigenetics, and the developing brain.
Neuropsychopharmacology 40: 141-153.
359. Bakulski KM, Rozek LS, Dolinoy DC,
et al. (2012) Alzheimer's disease and environmental exposure to lead: the
epidemiologic evidence and potential role of epigenetics. Curr Alzheimer Res 9:
563-573.
360. Dauncey MJ (2013) Genomic and
Epigenomic Insights into Nutrition and Brain Disorders Nutrients 5: 887–914.
361. Liu J, Zhao SR, Reyes T (2015)
Neurological and Epigenetic Implications of Nutritional Deficiencies on
Psychopathology: Conceptualization and Review of Evidence. Int J Mol Sci 16:
18129-18148.
362. Day JJ, Kennedy AJ, Sweatt JD
(2015) DNA methylation and its implications and accessibility for
neuropsychiatric therapeutics.Annu Rev Pharmacol Toxicol 55: 591-611.
363. Reynolds GP, McGowan OO, Dalton CF
(2014) Pharmacogenomics in psychiatry: the relevance of receptor and
transporter polymorphisms. Br J Clin Pharmacol 77: 654-672.
364. Cacabelos R (Ed) 2012 World guide
for drug use and pharmacogenomics. Corunna: EuroEspes Publishing.
365. Cacabelos R,
Torrellas C (2015) Pharmacoepigenetics: Medical Epigenetics. (1st edn), Elsevier, USA.
366. Zanger UM, Schwab M (2013)
Cytochrome P450 enzymes in drug metabolism: regulation of gene expression,
enzyme activities, and impact of genetic variation. Pharmacol Ther 138:
103-141.
367. Kim IW, Han N, Burckart GJ, Oh JM
(2014) Epigenetic changes in gene expression for drug-metabolizing enzymes and
transporters. Pharmacotherapy 34: 140-150.
368. Zanger UM, Klein K, Thomas M, et
al. (2014) Genetics, epigenetics, and regulation of drug-metabolizing
cytochrome p450 enzymes. Clin Pharmacol Ther 95: 258-261.
369. Sze CI, Su WP,
Chiang MF, et al. (2013)
Assessing current therapeutic approaches to decode potential resistance
mechanisms in glioblastomas. Front Oncol 3: 59.
370. Smith DF (2013) Quest for
biomarkers of treatment-resistant depression: shifting the paradigm toward
risk. Front Psychiatry 4: 57.
371. Gupta S, Verma S, Mantri S, et al.
(2015) Targeting MicroRNAs in Prevention and Treatment of Neurodegenerative
Disorders. Drug Dev Res 76: 397-418.
372. Junn E, Mouradian MM (2012)
MicroRNAs in neurodegenerative diseases and their therapeutic potential.
Pharmacol Ther 133: 142-150.
373. Parsi S, Smith PY, Goupil C, et
al. (2015) Preclinical Evaluation of miR-15/107 Family Members as
Multifactorial Drug Targets for Alzheimer's Disease. Mol Ther Nucleic Acids 4:
256.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4
-
Table 5
-
Table 6
-
Table 7
-
Table 8
-
Table 9
-
Table 10
-
Table 11
-
Table 12
-
Table 13
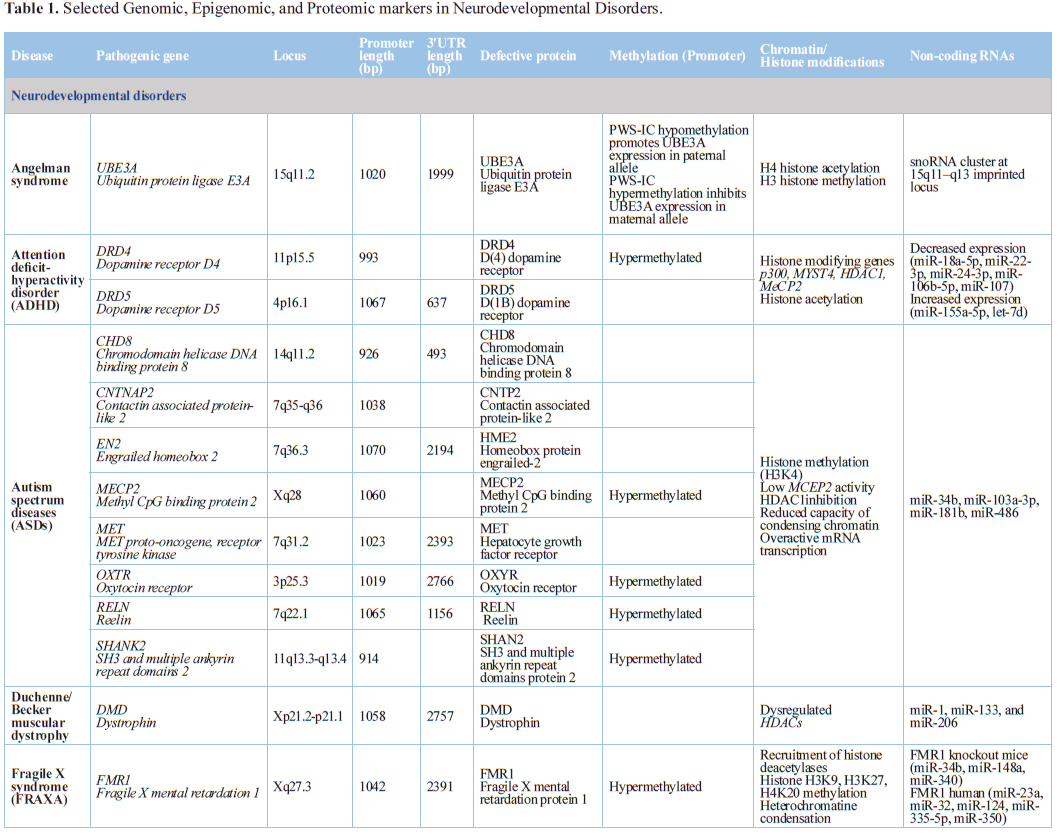
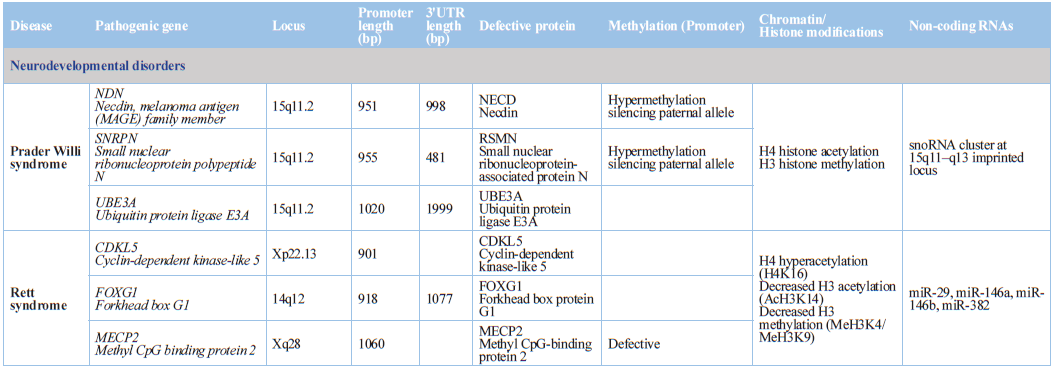
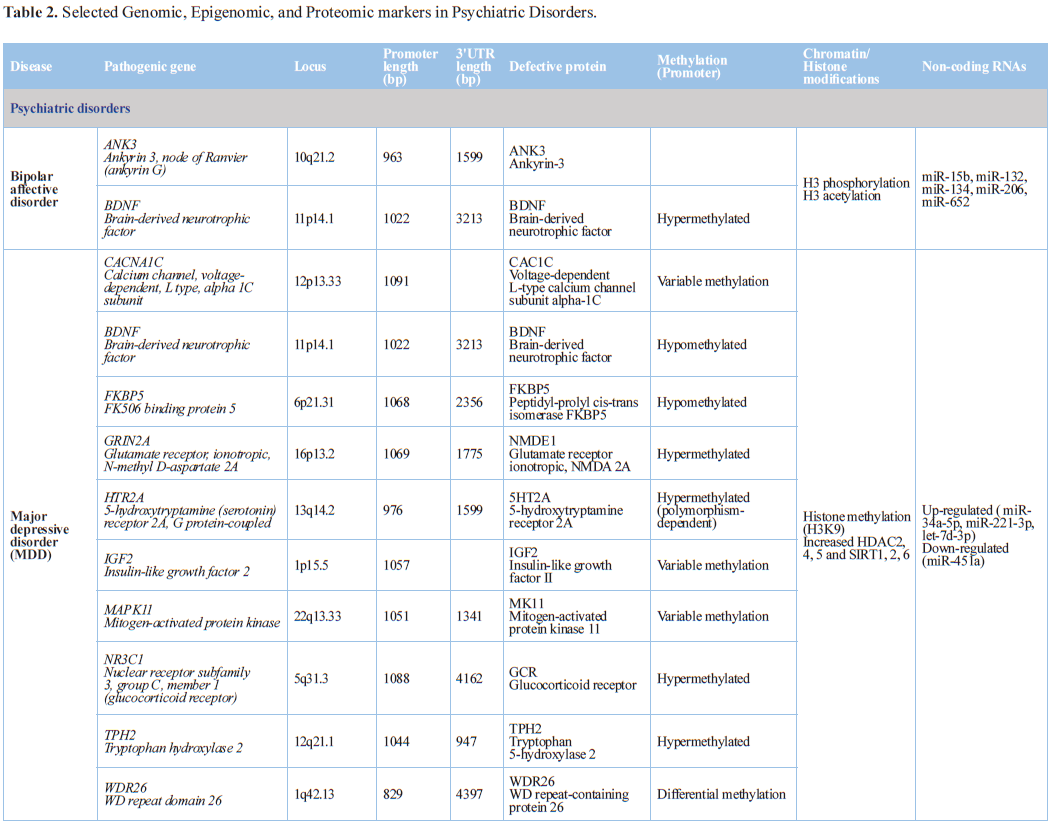
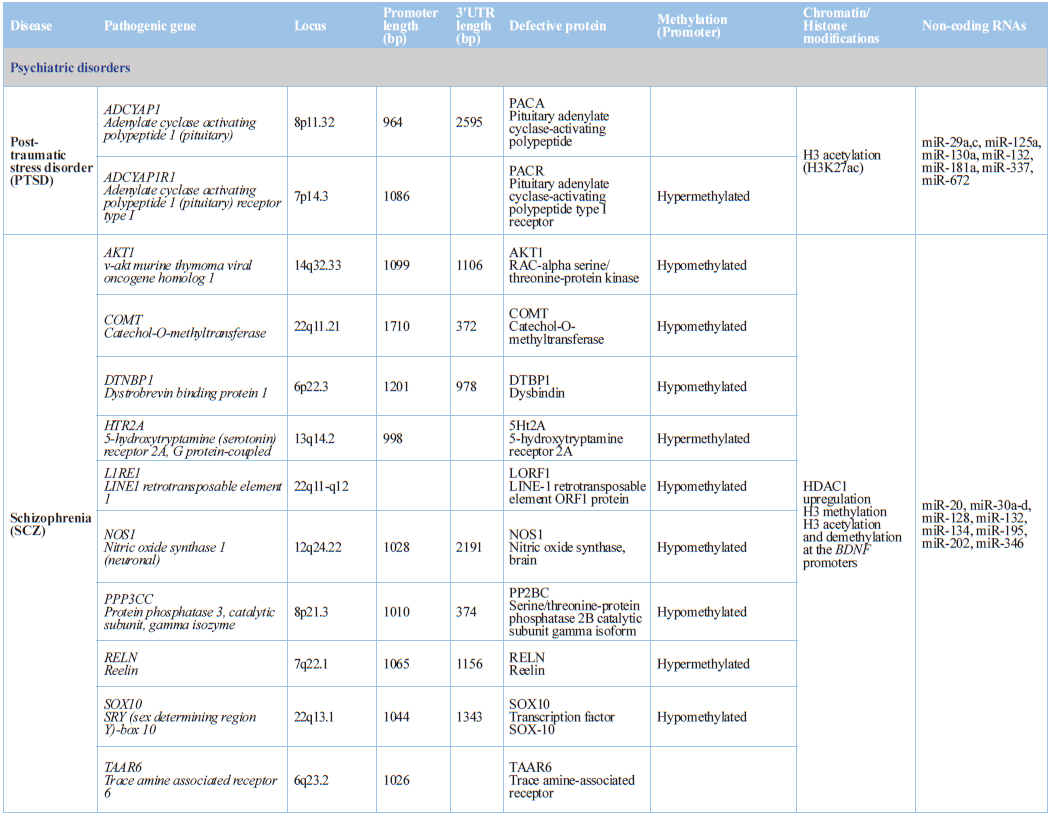
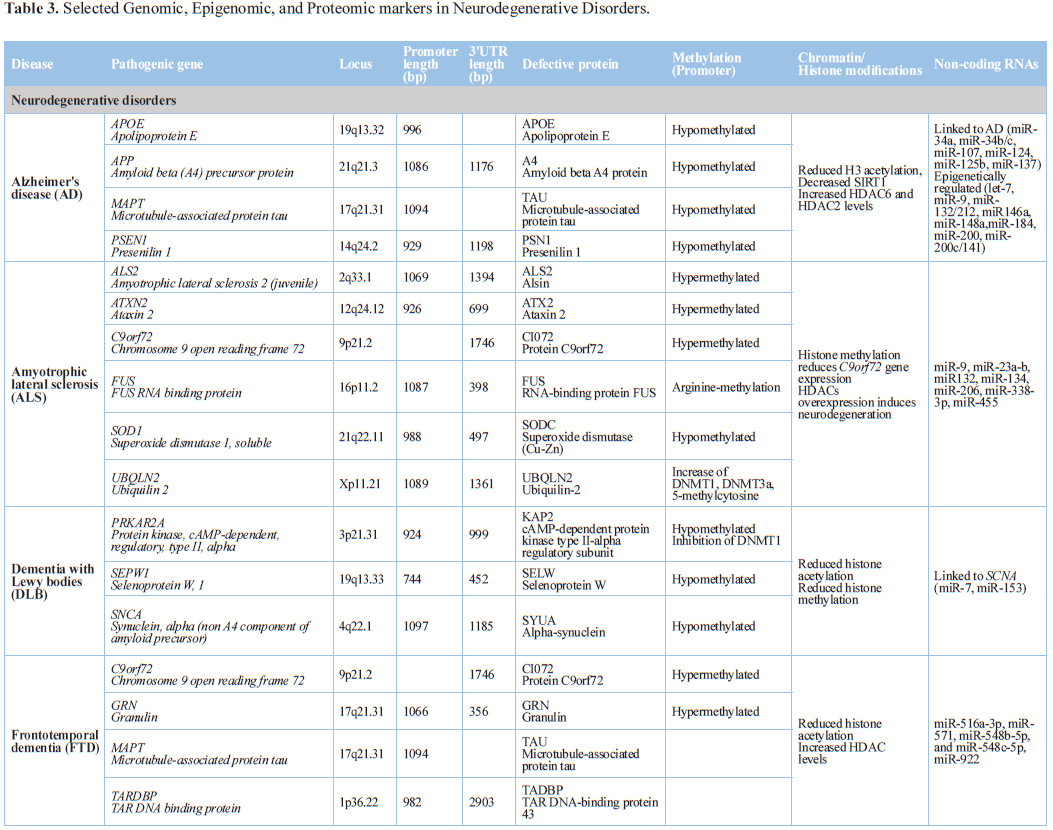
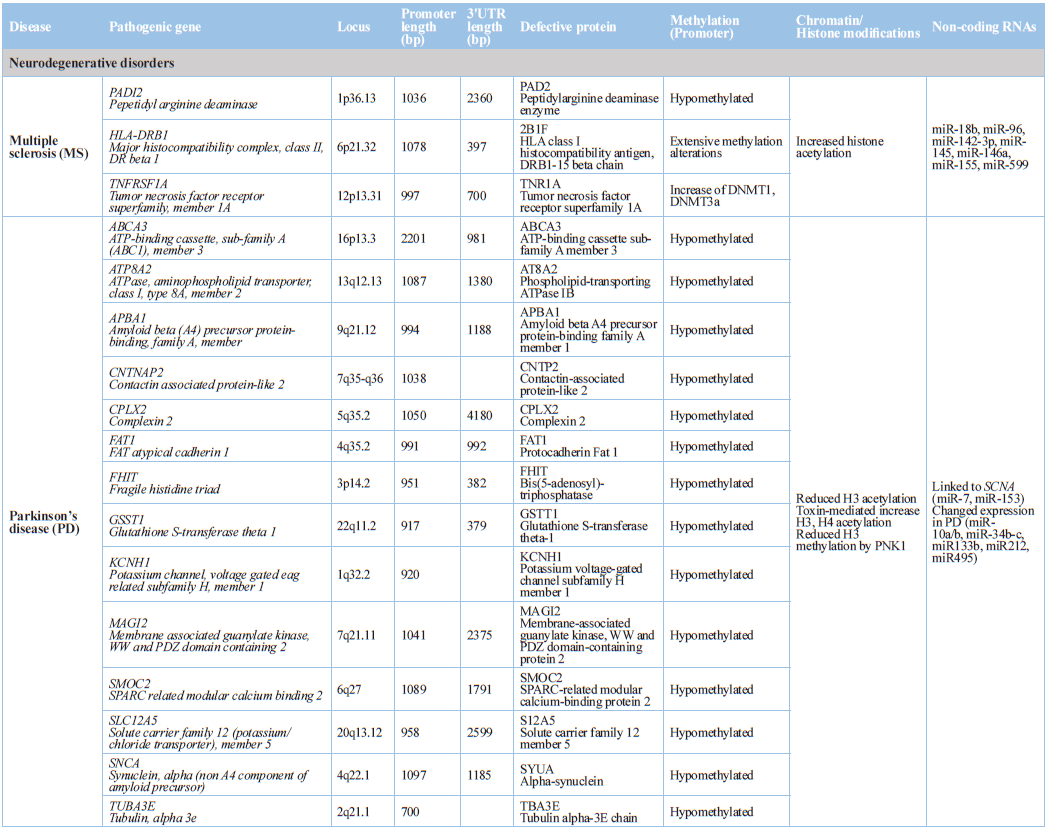
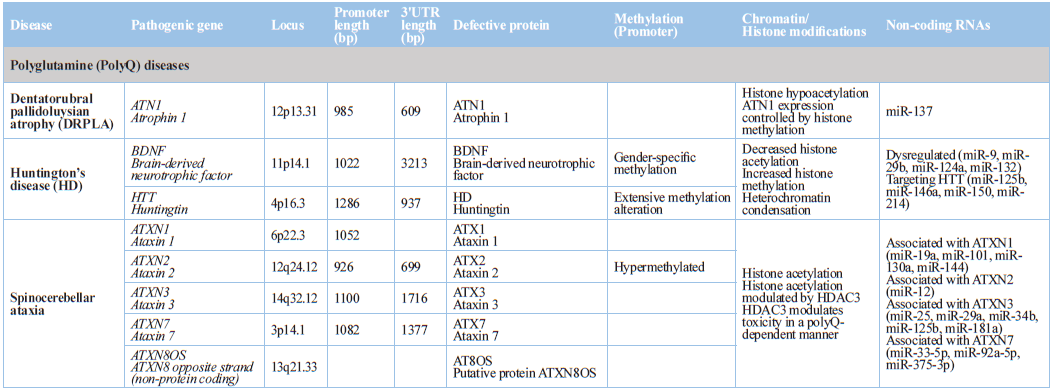
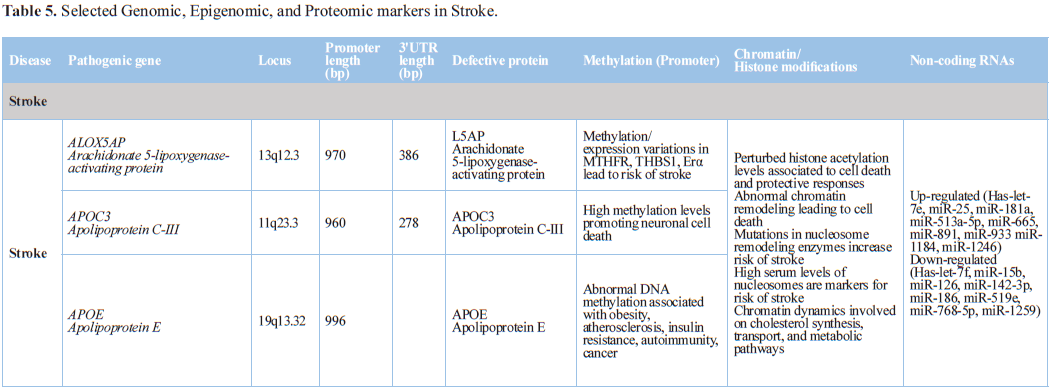
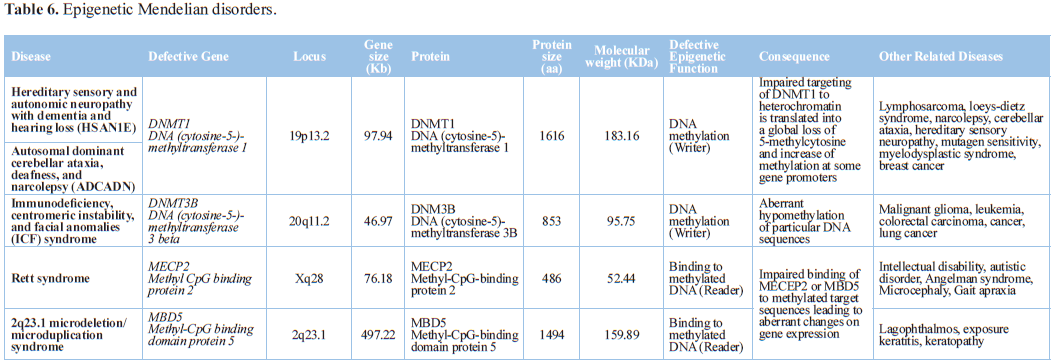
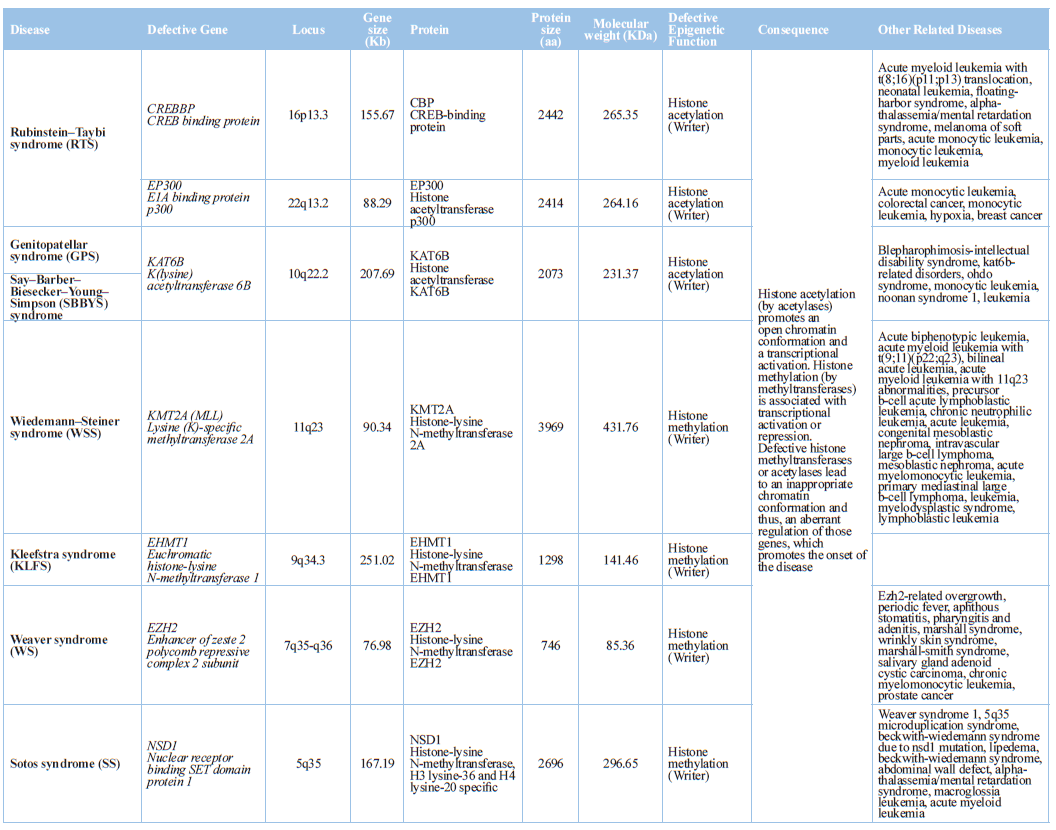
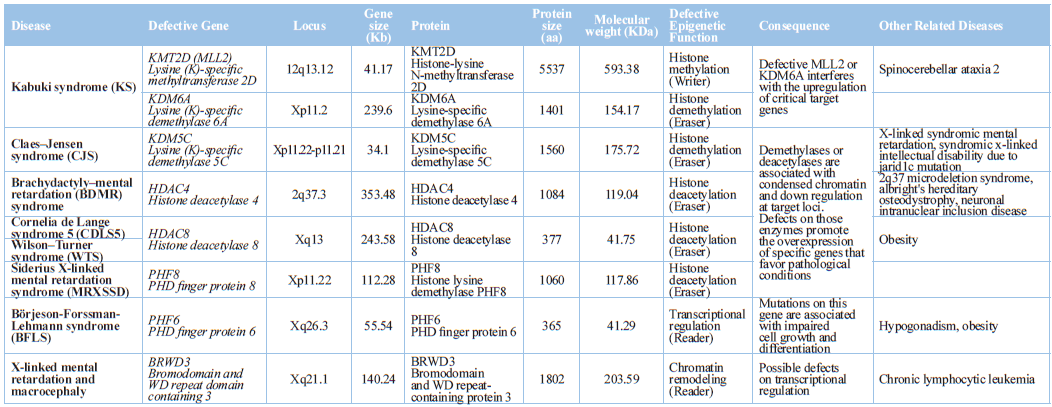
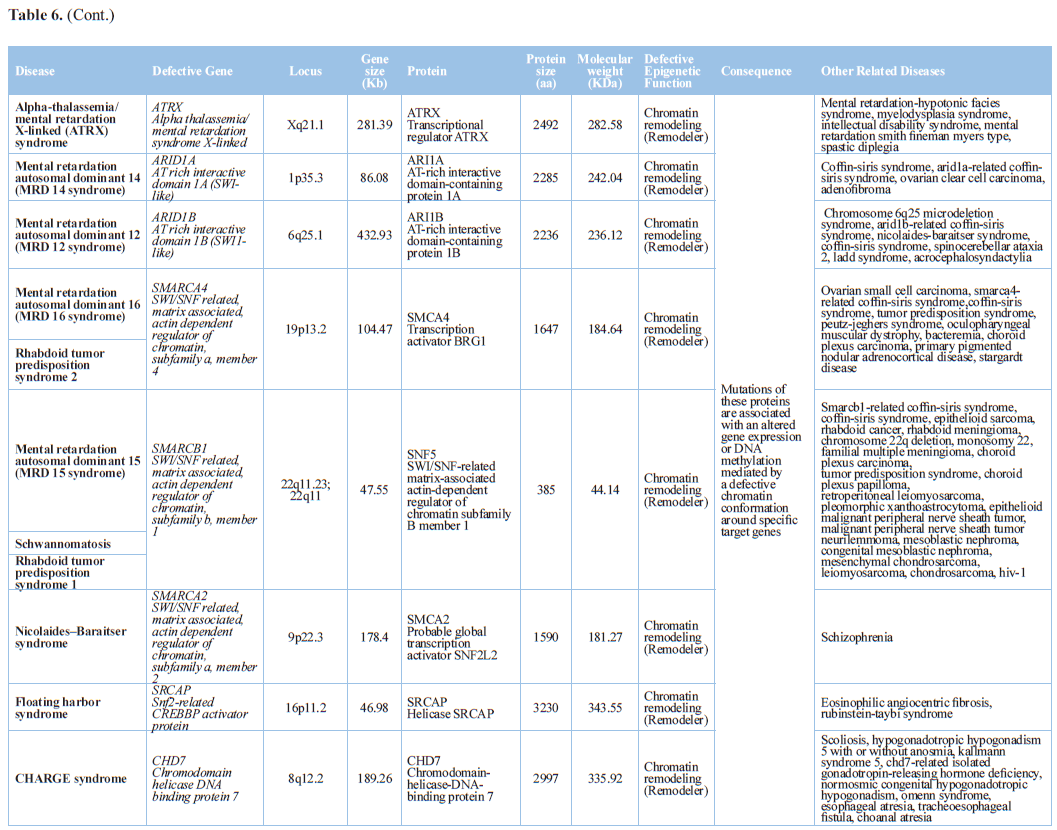
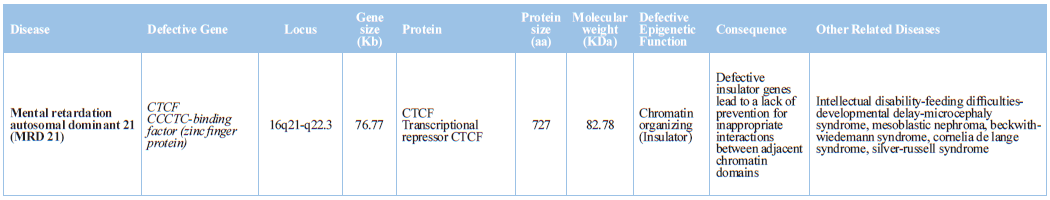
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Biochemistry and Molecular Medicine (ISSN:2641-6948)
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)
- Journal of Genetics and Cell Biology (ISSN:2639-3360)
- Proteomics and Bioinformatics (ISSN:2641-7561)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)
- Food and Nutrition-Current Research (ISSN:2638-1095)


